玉蝉颗粒制备工艺与质量标准研究
2022-04-01胡穗发刘建芳刘林林陈琳郭智强肖锐
胡穗发,刘建芳,刘林林,陈琳,郭智强,肖锐
1.南昌市洪都中医院,江西 南昌 330038;2.南昌市博泽康医药科技有限公司,江西 南昌 330029
咳嗽是呼吸系统疾病中常见的症状之一,中医学认为咳嗽既是肺系疾病中的一个症状,又是一种独立的疾病。《景岳全书》以简驭繁,将咳嗽分为外感咳嗽和内伤咳嗽两大类,一直沿用至今。中医治疗咳嗽可分为:(1)三因制宜,高度个体化,精准辩论治;(2)多环节,多靶点复方效应;(3)标本兼治[1]。咳嗽容易诱发支气管的痉挛,从而造成呼吸困难。蝉蜕具有散热、止咳平喘的作用,对表现为发热、咳嗽、咽痒等症可用蝉蜕辅助治疗。有研究证明蝉蜕提取物对支气管和肺组织炎性都有明确的改善作用,可通过改善血瘀情况来缓解支气管痉挛[2]。玉蝉颗粒由蝉蜕、芦根、桔梗等11 味中药组成,临床用于主治风邪犯肺型咳嗽[1]。玉蝉汤源于《温病条辩》中沙参麦冬汤和《伤寒论》的桔梗汤加减演化而来。在南昌市洪都中医院(以下简称我院)临床用于治疗咳嗽效果显著,无不良反应。现我院将该方制成中药颗粒剂,具有储存携带方便,质量稳定,广泛使用等优点。参考相关文献,对玉蝉颗粒的制备工艺和质量标准作如下研究。
1 仪器及材料
PWN225DZH 电子天平(美国奥豪斯仪器有限公司),YJDJ08 自动煎药包装机(长沙市卓成医疗器械有限公司),DZ-2BC 真空干燥箱(天津泰斯特仪器有限公司),1260 安捷伦高效液相色谱仪,超声仪KQ2200DB(昆山超声波仪器有限公司),NC-BY30 超纯水发生器(重庆隆暾科技有限公司),薄层层析用硅胶G 板(青岛海洋化工有限公司)。
麦芽糊精(批号:20220502M,江西景汉药用辅料公司),牛蒡苷对照品(批号:P2378091,上海诗丹德生物技术有限公司),百部对照药材(批号:121588-201502)、牛蒡子对照药材(批号:120903-201811)、甘草对照药材(批号:121303-201704)、桔梗对照药材(批号:121028-202113)、甘草苷对照品(批号:111610-201908)、缬氨酸对照品(批号:140681-2017803)、丙氨酸对照品(批号:140680-202005)均来源于中国食品药品检定研究院。色谱级乙腈、甲醇(购于安徽天地高纯溶剂有限公司),其他试剂为分析纯。玉蝉颗粒供试品及阴性样品由我院制剂室自制。
2 方法与结果
2.1 浸膏制备工艺优选
2.1.1 水提工艺优选以药材提取时的加水量、提取时间、提取次数为单因素指标,考察浸膏中牛蒡苷的含量,确定每个单因素的3 个水平,选定的因素水平见表1。再按玉蝉颗粒的处方比例,分别称取1 日剂量的饮片9 份,进行正交试验,并测定各试验组中牛蒡苷的含量。结果见表2。
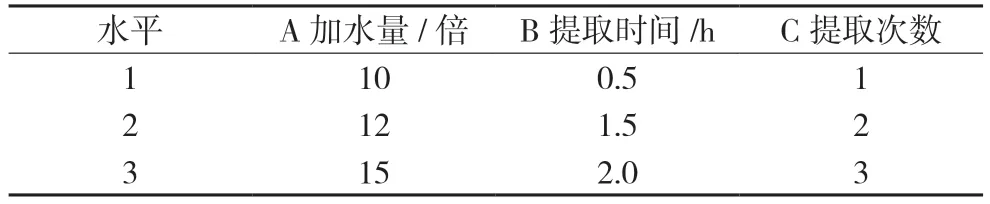
表1 正交试验因素与水平表
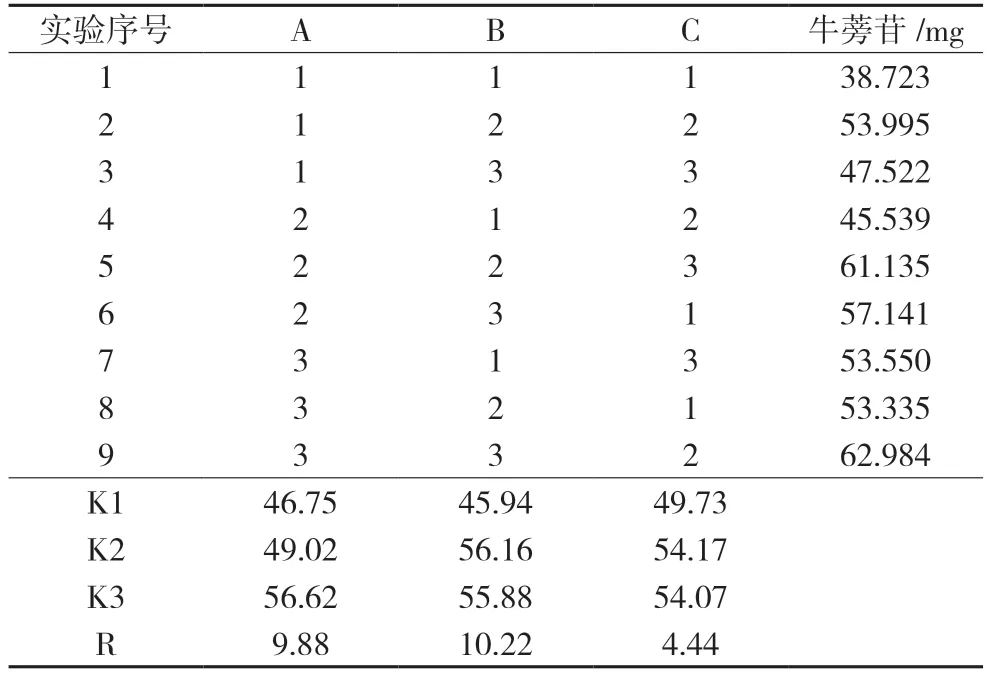
表2 正交试验结果及直观分析结果
由表2 直观分析结果可见,以牛蒡苷含量为评价指标考察提取效果的因素顺序为B>A>C,优选工艺为A3B2C2,即药材饮片加15 倍量水,煎煮两次,每次1.5 h。
2.1.2 浸膏得率验证试验根据优选的提取工艺条件A3B2C2,取玉蝉颗粒的处方比例饮片共3 份,进行平行验证试验,合并煎液,浓缩,65 ℃真空干燥成干膏。干膏得率结果见表3。3 次测定的平均值为24.20%,RSD 为1.18%,表明该工艺稳定可行。
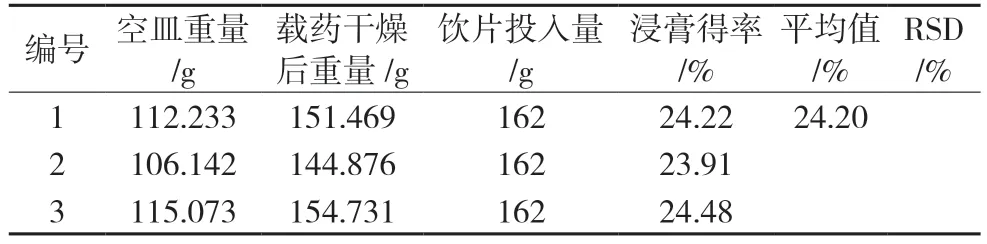
表3 浸膏得率结果
2.2 样品的制备
中药浸膏粉黏性大,且吸湿性强,直接制成颗粒存在困难,在浸膏粉中加入合适的辅料可以降低浸膏粉的黏性,同时也可改善其吸湿性和增加颗粒的流动性,对于颗粒剂的成型、分装与储存非常有利。颗粒剂传统制备时选用辅料有糊精、蔗糖、麦芽糊精、玉米淀粉等。麦芽糊精溶化性好、价格便宜,且在颗粒剂制剂中应用非常广泛,因此选用麦芽糊精作为辅料[3]。通过制粒难易来优选浸膏与麦芽糊精的比例,最终确定玉蝉颗粒中干膏粉:麦芽糊精按1∶0.7 的比例进行混合均匀后,以95%的乙醇为润湿剂制粒,70 ℃干燥2 h,即得。
2.3 薄层色谱鉴别
2.3.1 桔梗取玉蝉颗粒5 g,研细,加甲醇30 mL,超声处理30 min,滤过,滤液蒸干,残渣加水30 mL溶解,用水饱和正丁醇60 mL 振摇提取,分取正丁醇液,用氨试液60 mL 洗涤,弃去氨液,取正丁醇液蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液。另取桔梗对照药材1 g,加50%甲醇50 mL,同法制成对照药材溶液[4]。采用硅胶G 薄层板,点样量各5 μL,以三氯甲烷-甲醇-水(7∶3∶0.5)为展开剂,展开,取出,晾干,喷以5%硫酸乙醇溶液,在105 ℃加热至斑点显示清晰。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性无干扰。见图1。

图1 桔梗薄层色谱图
2.3.2 百部取玉蝉颗粒1.5 g,研细,加乙醇20 mL和盐酸1 mL,加热回流30 min,滤过,滤液蒸干,残渣加甲醇2 mL 使溶解,作为供试品溶液。取百部对照药材0.5 g,加水50 mL,加热回流30 min,滤过,滤液蒸干,残渣自“加乙醇20 mL 与盐酸1 mL”起,同法制成对照药材溶液[5]。采用硅胶G薄层板,点样量各1 μL,以甲苯-乙酸乙酯-甲酸(4∶2∶0.5)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性无干扰。见图2。

图2 百部薄层色谱图:1.百部对照药材;2~4.玉蝉颗粒供试品;5.阴性样品
2.3.3 牛蒡子取玉蝉颗粒10 g,研细,加水50 mL溶解,加乙醇至含醇量60%,静置,滤过,滤液蒸干,残渣加水20 mL 溶解,用水饱和的正丁醇振摇提取3 次,每次20 mL,合并正丁醇液,用氨试液40 mL洗涤,弃去氨液,取正丁醇液蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液。另取牛蒡子对照药材1 g,加乙醇20 mL,加热回流1 h,滤过,滤液浓缩至约2 mL,作为对照药材溶液[6]。采用硅胶G 薄层板,点样量各2 μL,以三氯甲烷-甲醇-水(40∶10∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显示清晰。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性无干扰。见图3。
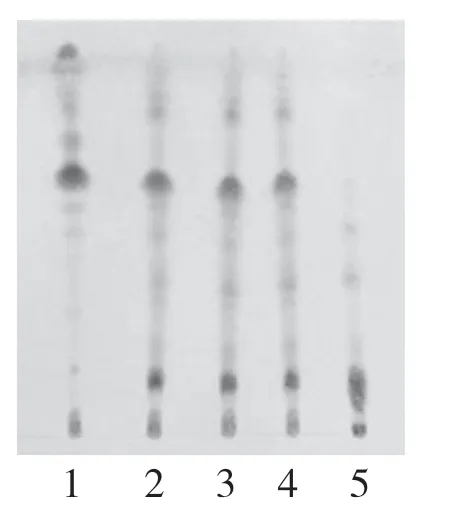
图3 牛蒡子薄层色谱图:1.牛蒡子对照药材;2~4.玉蝉颗粒供试品;5.阴性样品
2.3.4 炙甘草取玉蝉颗粒3 g,研细,加甲醇30 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加水40 mL 溶解,用正丁醇60 mL 振摇提取,分取正丁醇液,用水60 mL 洗涤,弃去水洗液,取正丁醇液蒸干,残渣加甲醇10 mL 使溶解,作为供试品溶液。另取甘草苷对照品,加甲醇制成每1 含1 mg 的溶液,作为对照品溶液[7]。采用硅胶G 薄层板,点样量各1 μL,以乙酸乙酯-甲酸-冰乙酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干喷以10%硫酸乙醇溶液,在105 ℃加热3 min,立即置紫外光灯(365 nm)下检视。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性无干扰。见图4。

图4 甘草薄层色谱图:1.甘草苷对照品;2~4.玉蝉颗粒供试品;5.阴性样品
2.4 含量测定方法与结果
2.4.1 液相色谱条件[8]选用Agilent 5 TC-C18 色谱柱(250 mm×4.6 mm,5 μm);以甲醇为流动相A,以磷酸溶液(pH=2.58)为流动相B,梯度洗脱比例按表4进行;检测波长280 nm;流速为1.0 mL/min;柱温30 ℃。

表4 梯度洗脱表
2.4.2 对照品溶液制备取牛蒡苷对照品,精密称定,加甲醇制成每1 mL 含36 μg 的溶液,即得。
2.4.3 供试品溶液制备取玉蝉颗粒粉末约5 g,精密称定,置锥形瓶中,精密加入甲醇50 mL,密塞,称定重量,超声处理60 min,放冷,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液25 mL,蒸干,残渣加水30 mL 使溶解,用乙酸乙酯萃取6 次,每次30 mL,合并乙酸乙酯层,置水浴蒸干,残渣加甲醇使溶解并定容至25 mL,摇匀,即得。
2.4.4 阴性对照溶液的制备按样品制法制备缺牛蒡子的阴性样品,取适量,按供试品溶液的制备方法制备阴性对照溶液。
2.4.5 专属性考察分别吸取对照品溶液、供试品溶液与阴性对照溶液各5 μL,按“2.4.1”项下的色谱条件测定,记录高效液相色谱图,结果方法专属性好,阴性对照无干扰。见图5。
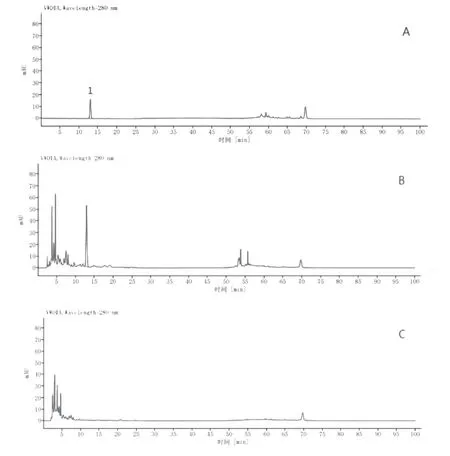
图5 HPLC色谱图:A.对照品溶液;B.供试品溶液;C.阴性对照溶液
2.4.6 提取方式、提取时间考察采用加热回流提取和超声提取分别对样品进行处理并测定含量,当提取方式为超声提取,提取时间为60 min 时,牛蒡苷含量最高。见表5。

表5 提取方式与提取时间试验结果
2.4.7 线性关系考察以甲醇为稀释剂对牛蒡苷对照品储备液(0.719 1 mg/mL)进行稀释,制备浓度分别为0.071 9 mg/mL、0.215 7 mg/mL、0.251 7 mg/mL、0.431 5 mg/mL、0.503 4 mg/mL、0.719 1 mg/mL 的对照品溶液,按“2.4.1”项下色谱条件测定。得峰面积(Y)-浓度(X,mg/mL)线性回归方程为:Y=2 817.1X-6.299 2,r=0.999 7,试验结果表明牛蒡苷在0.071 9 mg/mL~ 0.719 1 mg/mL 浓度范围内呈良好线性关系。
2.4.8 精密度试验精密吸取浓度为0.359 6 mg/mL的牛蒡苷对照品溶液5 μL,按“2.4.1”项下色谱条件测定,记录峰面积,重复进样6 次。牛蒡苷峰面积RSD 为0.18%,表明仪器精密度良好。
2.4.9 溶液稳定性试验取同一供试品溶液按“2.4.1”项下色谱条件,分别于室温放置0 h、3 h、8 h、15 h、23 h、27 h、36 h 后测定峰面积,结果RSD 为0.71%,说明样品溶液在36 h 内稳定
2.4.10 方法耐用性试验更换不同的操作人员、不同色谱柱和不同高效液相色谱仪进行方法的耐用性考察,结果该方法稳定耐用。
2.4.11 重复性试验按“2.4.3”项下的方法平行制备6 份供试品溶液,按“2.4.1”项下色谱条件测定,结果测得样品中牛蒡苷平均含量为5.191 3 mg/g,RSD 为1.32%,表明该方法重复性良好。
2.4.12 回收率试验取已知含量的玉蝉颗粒样品粉末9 份,每份约2.5 g,精密称定,每3 份为一浓度组,分别加入低、中、高浓度的牛蒡苷对照品,按“2.4.3”项下方法制得回收率供试溶液品。分别吸取5 μL注入液相色谱仪,计算牛蒡苷的回收率,结果平均加样回收率为95.70%,RSD 为0.51%,表明该方法准确性良好。见表6。
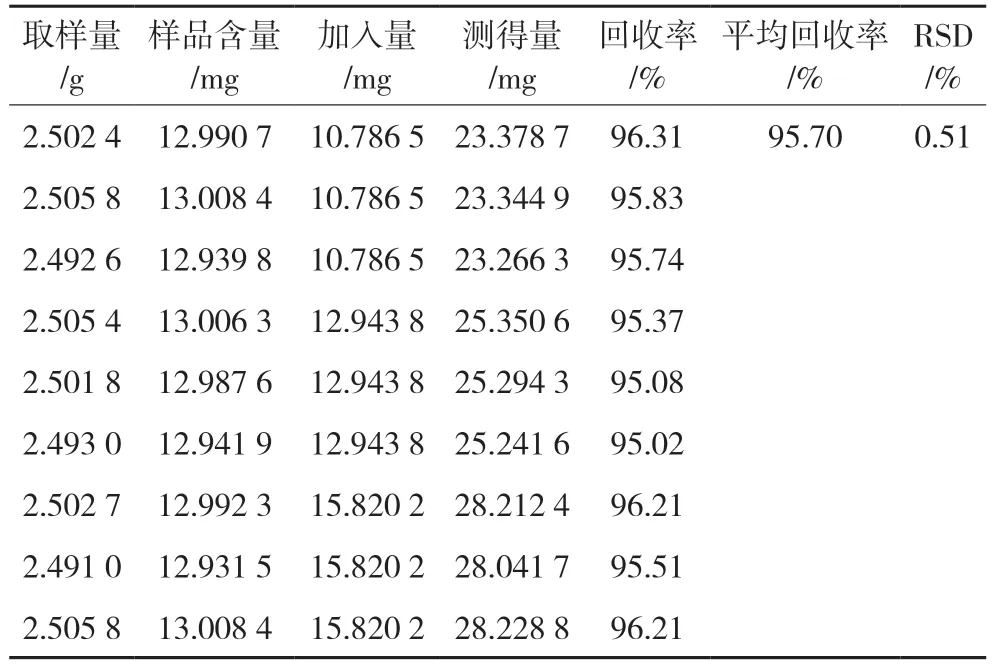
表6 加样回收试验结果
3 讨论
玉蝉颗粒重用蝉蜕,其水溶性成分为氨基酸。氨基酸HPLC 含量测定需要柱前衍生成具有荧光吸收的物质,经过氨基酸专用分析色谱柱洗脱在紫外可见光下进行定量[9]。柱前衍生对实验环境和实验者要求较高,操作过程不易控制。且氨基酸的专属性不强,故本研究未纳入蝉蜕的含量测定,期待后续能完善方法。
玉蝉颗粒采用了薄层色谱方法对桔梗定性鉴别。桔梗皂苷的紫外最大吸收波长为205 nm,属末端吸收,溶剂干扰较大,桔梗皂苷E 和D 含量低,单点外标法测定误差较大,故未将桔梗皂苷含量测定纳入本研究。
本研究采用正交试验优化了提取工艺并优选了制粒所用辅料和方法。建立了处方中桔梗、百部、牛蒡子、炙甘草四味药材的薄层色谱鉴别和牛蒡苷的高效液相色谱含量测定方法。所建立的方法为更好地控制该玉蝉颗粒的质量提供了依据。
