肥厚型心肌病基因型–表型关联研究进展
2022-03-26舒甜胡豪畅沈才杰林少沂陈晓敏
舒甜,胡豪畅,沈才杰,林少沂,陈晓敏
综 述
肥厚型心肌病基因型–表型关联研究进展
舒甜1,2,胡豪畅1,2,沈才杰1,林少沂1,陈晓敏1,2
1. 浙江大学宁波医院心血管内科,宁波 315000 2. 浙江大学医学院,杭州 310029
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一种以左心室肥厚为突出特征的常染色体显性遗传病,其发病率为1/500~1/200。目前已发现超过30个基因的1500种突变与该疾病的发生发展相关,致病基因变异连同修饰基因多态性、环境因素等影响因素发挥作用,使得疾病表型极具异质性,临床表现上从无任何症状到心源性猝死均可发生,病理表型主要包括心肌细胞肥大、排列紊乱及纤维化、心肌缺血等。近年来,许多研究致力于探究HCM基因型对表型的影响, 并基于遗传背景对HCM的治疗方法进行研发。本文以HCM基因型–表型的关联为重点,从HCM的致病基因、关联影响因素和最新治疗手段等多方面综述了HCM的研究进展,以期为研究HCM的发生发展及治疗方向提供遗传学方面的思路。
肥厚型心肌病;致病基因;表型;治疗
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是最常见的遗传性心脏病,也是青少年运动员猝死的首要原因。在过去的20年里,研究表明HCM的全球发病率为1/500,估算我国的成年HCM患者超过100万人[1],但随着医学影像技术的进步以及人们对健康体检的愈发重视,近来的研究显示HCM发病率已增至1/200[2],也提示了我国HCM患者的数量远不止100万。部分HCM患者终生没有任何症状,但也有一部分HCM患者面临重度心力衰竭、严重心律失常甚至心源性猝死的境况。因此,为了尽可能地减少疾病风险,早期识别HCM高危人群、把握HCM的发生发展规律至关重要。
现今已发现超过30个基因的1500种突变与HCM发病相关,以编码肌小节蛋白的基因突变为主[2]。HCM的管理指南推荐对患者的致病基因进行遗传检测,用于做出精确诊断和早期筛查高危患者[1,2],但由于缺乏HCM基因型和表型的关联分析,加上HCM的基因型和表型都有很大的异质性,借助基因型来预测疾病走向的应用十分受限。为突破这一壁垒,近年来许多研究对HCM患者或是携带致病基因变异的患者家属进行长期随访,结合基因型对研究人群的临床症状和预后进行分析,使得HCM基因型–表型的关联更加清晰和丰富。本文在介绍HCM致病相关基因和表型的基础上,重点讨论了HCM基因型–表型关联以及这种关联的影响因素,并结合遗传背景综述了HCM在治疗手段方面的最新进展,以期为HCM的诊治提供更多遗传学依据。
1 HCM的致病基因
作为遗传性疾病,目前已有8个编码肌丝蛋白的基因被证明为HCM的致病基因,包括编码粗肌丝蛋白的、、、和编码细肌丝蛋白的、、、[3,4]。和是最为多见的致病基因,分别导致了50%和30%~35%的HCM。除了肌丝蛋白基因外,也有很多编码非肌丝蛋白的致病基因被报道,包括编码Z盘的基因、编码肌联蛋白的基因、编码细胞骨架蛋白的基因[5,6]和编码钙调节处理蛋白的基因[7~13](图1)。其中编码细胞骨架蛋白的致病基因约占1%~2%,编码钙调节处理蛋白的致病基因较为罕见。
随着基因检测的普及,越来越多的研究对HCM患者进行了基因检测和数据分析。先前研究显示不同种族的HCM患者基因突变位点可能不同[14]。北京阜外医院宋雷课题组[15]通过对常见变异进行关联分析发现变异位点和HCM相关并特异存在于东亚人群中,这提示不同种族在遗传背景上存在差异。因此,在研究致病基因特点时,需考虑到种族和区域的差异。
2 HCM的表型
HCM的形态学、组织学特征和临床表型是多种因素共同作用的结果,因此HCM患者的表型特征具有多样性。HCM的左心室肥厚可发生在心室游离壁、心尖部、乳突肌等多个位置,最为典型的是发生在室间隔基底部,易引起左心室流出道梗阻。HCM的基本组织病理学特征为心肌细胞肥大和排列紊乱,具体程度因人而异,部分患者还会出现不同发展程度的心肌纤维化和/或微循环障碍。
HCM的典型特征是心室收缩功能增强、舒张功能减退,或合并有心肌缺血、心律失常、心力衰竭等并发症。临床表型上,HCM患者可表现为呼吸困难、心悸、胸痛、头晕、晕厥、心源性猝死等。需要注意的是,HCM的症状并不一定和心室肥厚及流出道梗阻程度相关,甚至部分患者始终没有出现临床症状。由此可见,HCM 具有极大的临床异质性,分析HCM的基因型和表型的相关性对于尽早识别HCM患者、分析患者病情及改善患者预后具有重要意义。
3 HCM的致病基因–表型关联
国内外学者对先证者及家属进行基因检测和随访发现,部分携带致病基因变异的人群并不发病,由此对疾病外显率展开了探究。先前几项研究对未发病的携带者进行了5~10年的随访,获得的外显率均不超过18%[16~20]。而最新一项大型研究对285位携带者进行了15年的随访并发现外显率为56%,显著高于先前的研究[21]。和突变的发病风险最高,分别为43%和66%,而这两种基因突变在该研究中的占比较大,加上随访年限的延长,可能是导致该项研究中外显率提高的原因。
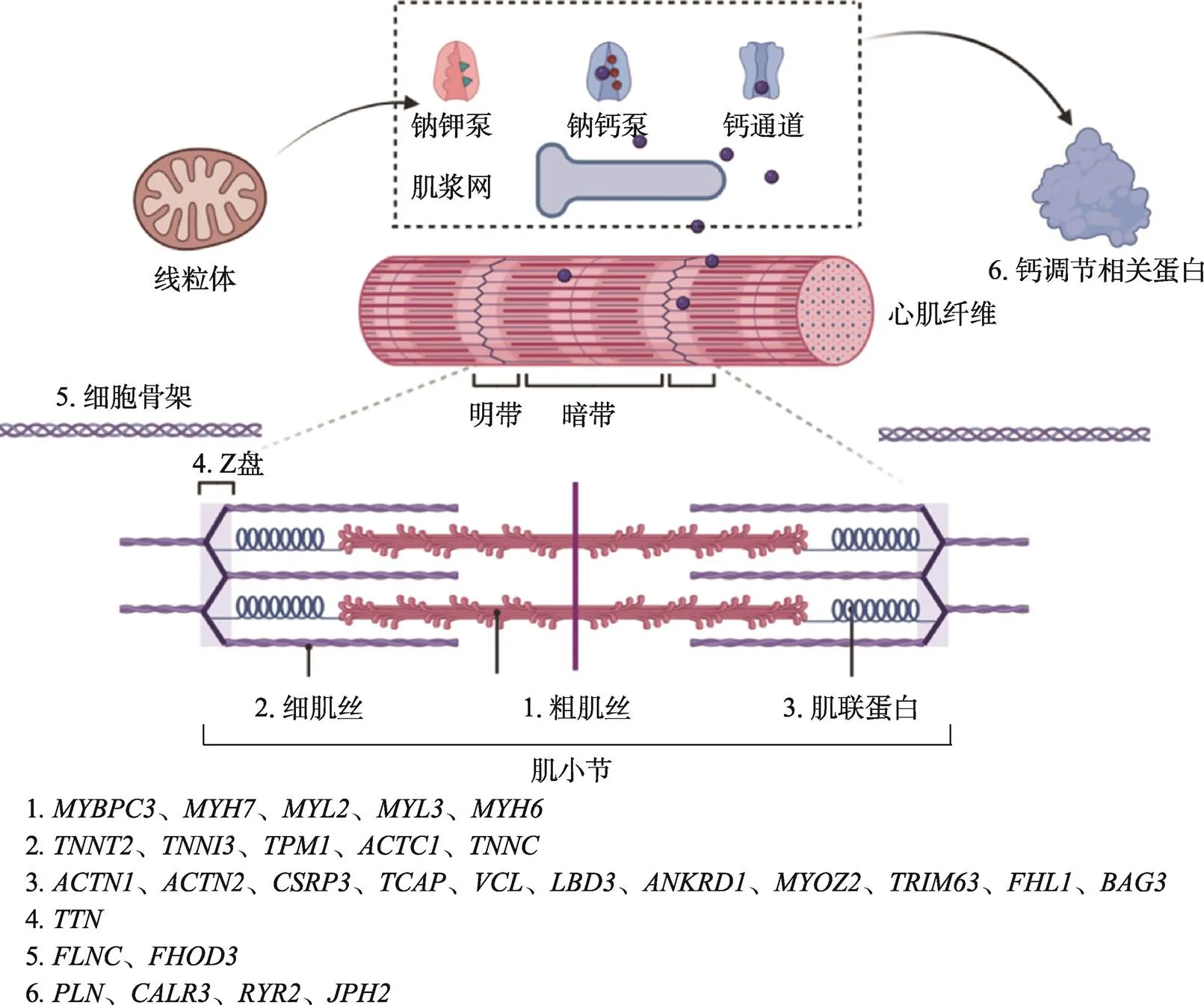
图1 心肌细胞内各结构上存在的致病基因
肌节是心肌收缩的基本单位,主要由粗肌丝和细肌丝组成,粗细肌丝间的相互滑动产生了肌节的收缩。肌节单位之间由Z盘连接,Z盘是肌节收缩的着力点。肌联蛋白连接了粗肌丝和Z盘,在心肌收缩和舒张中起到了分子弹簧的作用。细胞骨架对维持肌节结构和功能起到重要作用,而钙调节相关蛋白则通过收缩耦联机制影响心肌收缩舒张功能,因此编码粗肌丝的、、、、,编码细肌丝的、、、、,编码Z盘的、、、、L、、、、、、,编码肌联蛋白的,编码细胞骨架、和编码钙调节相关蛋白的、、、均可能为HCM的致病基因。
在确诊为HCM的患者中,60%的患者携带致病基因变异,其余40%的患者可能具有新的基因变异或为“非家族性HCM”患者(即不具有致病基因变异,且无HCM家族史的患者)。对HCM患者的心肌标本进行病理分析发现,心肌细胞排列紊乱[22]、微血管功能损伤及心肌纤维化程度在携带致病基因变异的患者中更为严重,这类患者的室间隔厚度更大,形态多表现为反向曲度,而未携带致病基因变异的患者多具有孤立性室间隔基底部肥大。此外,携带致病基因变异的患者更早发病,进展为房颤、心衰甚至发生心源性猝死的风险更高[23,24]。由此不难发现,携带致病基因变异的患者在病理结构及临床预后方面的表现更差。
3.1 粗细肌丝基因–表型关联
HCM的致病变异大多发生在编码肌小节粗细肌丝蛋白的基因中。细肌丝基因变异通常引起心尖肥厚型和向心型HCM,这两种心室形态一般不会造成流出道梗阻,引起室壁增厚的程度也较轻。而粗肌丝基因突变引起室间隔型HCM较多,发病早,心室肥厚和流出道梗阻更严重,对侵入性治疗的需求更大[25]。一项纳入了7675例患者的Meta分析结果验证了粗肌丝基因变异会导致更严重的心肌肥厚和梗阻这一观点,但发现细肌丝基因变异更易促发心衰进展[23]。
在粗肌丝基因中,和变异是最早被发现且占比最多的致病基因变异。变异主要是移码突变,通过无义介导的mRNA衰变引起单倍体不足、产生毒性多肽,导致肌球蛋白结构功能异常。致病机制较复杂,除了通过改变肌球蛋白头上ATP酶的活性来加强肌球–肌动蛋白相互作用外,还会涉及能量代谢异常、纤维化、钙超载等多种机制。在有HCM家族史的儿童中,、变异引起早发HCM的风险最大,分别是未携带致病基因变异队列的3.2、2.3倍,并易引发心血管不良事件[25]。在病理和临床表型上,现在普遍认为的“恶性”程度高于,即携带致病变异的HCM患者发病更早,室间隔厚度更大,心室收缩及舒张功能障碍更严重,引起传导阻滞、房颤、室性心律失常等并发症的风险增加[23,26,27]。我国关于、的报道相对较少,根据现有的少量病例报告显示,突变和房颤、心力衰竭相关[28]。
细肌丝的肌钙蛋白C和钙离子结合,原肌球蛋白发生位移,暴露出细肌丝上和横桥结合的位点。这类致病变异钙依赖性地增强了粗细肌丝的结合,引起心肌细胞过度收缩。突变是最多见的致病性细肌丝基因变异。国外研究发现,携带突变的HCM患者发生心源性猝死的风险较大,但国内一项研究结果却表明,27名携带该基因突变的HCM患者发生心血管死亡的风险和对照组相似[29],说明基因型–表型关联在不同种族之间可能存在差异。突变在临床上并不多见,与较高的外显率及严重的疾病表型相关[30]。突变发病风险相对较低,缺乏足够样本量的预后分析。尽管上述变异都发生在细肌丝基因中,但它们引起的临床表型并不一致。近期还有研究表明,同样是第92位氨基酸发生了变异,和临床症状和对地尔硫卓的治疗反应却存在差异[31],说明了同一位点发生突变所产生的HCM表型也不尽相同。
3.2 其他基因–表型关联
除了粗细肌丝基因外,编码非肌小节蛋白的基因变异也对HCM的表型起重要调控作用。Z盘是心肌细胞的信号转导中心,是肌丝收缩的着力点。在编码Z盘相关蛋白的基因中,携带变异的HCM患者病程发展迅猛,可能是需要预先植入复律器的潜在高危人群。而杂合变异携带者的HCM在心室肥厚程度上存在较大差异,一般表现为低风险形式,但也有心源性猝死的报道[32,33]。钙调节相关蛋白基因变异会影响心肌细胞Ryanodine(RyR)受体、肌浆网钙ATP酶(SERCA)的活性,在收缩期触发肌浆网释放大量钙,减少舒张期钙的回收。携带这类突变的HCM患者具有较轻的梗阻程度和低猝死风险,但需着重防范心律失常的发生[33,34]。
和是编码细胞骨架蛋白的基因。编码丝状蛋白C,该基因发生致病变异可使肌动蛋白形成聚集体,损害肌节功能,导致HCM的发生。在发现该突变的9个家系中,5个家系有心源性猝死的患者,表明该基因突变可能和高风险猝死相关[6]。在心脏中表达,能够和肌动蛋白结合,辅助心肌收缩,该基因发生突变是HCM患者心血管死亡的独立危险因素[5]。由于上述基因和表型之间的关联大多基于小样本量甚至个别病例报道,仍需进行大量功能实验明确其致病性。
部分研究提示一些患者携带2~3个致病变异,且具有多突变会增加室性心律失常和心力衰竭风险,临床预后明显不良[35]。总之,基因型对HCM患者表型具有重要作用,但这种作用仍需开展更多的基础及临床研究来加以发掘和验证,从而在指导临床诊疗上发挥更大的价值。
4 致病基因–表型关联的影响因素
近年来,人们发现一些携带同一致病变异的家族成员在心脏结构和临床表型上具有极大差异,证明除了致病基因的影响外,HCM表型还受其他影响因素的调控,包括修饰基因多态性和环境因素。
4.1 修饰基因的作用
RAS通路上的基因是HCM发生发展中至关重要的修饰基因。有研究表明,突变单发时预后较突变好,但合并血管紧张素转化酶(angiotensin converting enzyme, ACE)的等位基因变异时,疾病程度会加重。Meta分析显示的等位基因是遗传危险因素[36],在具有MYBPC3突变的基础上,携带/会导致发病年龄提早3~4年,室壁厚度进一步增大。血管紧张素原(angiotensinogen, AGT)基因多态性里,是一个关键位点,纳入8项研究的Meta分析表明隐性遗传模型(vs+)会提高HCM发病风险[37]。另有个别研究发现酶()多态性也会提高HCM外显率[38],携带和多态性的患者中具有左心室流出道梗阻和心衰表型的占比更大[39]。这些研究都能表明,在发生致病基因变异的基础上,RAS通路修饰基因多态性对发病和严重程度起到了重要的修饰作用。
肌球蛋白结合蛋白H(MYBPH)和肌联蛋白(titin)都是肌节的的组成部分,前者协同MYBPC发挥作用,调节肌节收缩;后者相当于分子弹簧,对粗、细肌丝进行精确的调控,在耦联和协调心肌的舒缩运动中发挥着重要作用。突变会增加携带的HCM患者的室间隔厚度[40]。等位基因能够通过无义介导mRNA使正常的肌原纤维减少,导致肌小节异常,最终破坏心脏收缩。我国有研究发现,可能作为修饰基因,使HCM心血管死亡事件的发生风险增加[41]。
临床研究表明编码电压门控L型钙通道β2 (L-type calcium channel β2, CACNβ2)的基因突变能够提高外显率,加重肥厚程度[42]。在涉及钠钙稳态通路蛋白的基因中,编码电压门控钠通道的发生异常剪辑会促进心律失常的发生[43]。
此外,编码细胞色素氧化酶P450、内皮素、一氧化氮合酶相关蛋白的基因都能起到修饰作用来促使心肌细胞肥大。已知的致病基因如,也可能作为修饰基因来调控疾病表型。今后,我们需要通过基础实验来明确修饰基因对致病基因–表型产生影响的具体机制。
4.2 环境因素的作用
在致病基因变异作用的基础上,除了修饰基因多态性,环境因素也会影响发病风险和病情程度。韩国一项全国性的随访研究将具有高脂血症、糖尿病或高血压定义为不健康的代谢状态,发现不健康代谢状态组在随访期间被诊断为HCM的风险比对照组高出1.5倍。多项队列研究探索了肥胖和HCM表型的关联,发现肥胖会影响HCM患者的心脏结构、临床表现和预后。左房内径、左室间隔厚度及心脏重量指数随着BMI分级的增加而增大,发生流出道梗阻可能性也增大。肥胖患者在NYHA分级、活动耐量上表现更差、出现房颤、心衰等不良结局的风险更大[44]。合并糖尿病的HCM患者在心脏结构及临床症状上的表现也更为严重,且15年死亡率比对照组升高了7%[45]。此外,高血压会作为危险因素增加携带突变的HCM患者的发病风险[28]。在肌小节致病基因变异阴性的患者中,舒张压每增加1个标准差,疾病风险增加4倍[46]。睡眠呼吸暂停综合征(obstructive sleep apnea,OSA)能够增加包括心衰、心肌梗死在内的许多心血管疾病的发病率及病死率,合并OSA的HCM患者有更为明显的心脏结构功能损伤,出现房颤的风险为不合并OSA的4倍[47]。此外,吸烟饮酒等不良生活方式、抑郁等情绪因素都可能对HCM表型产生不利影响。
随着环境因素的作用得到了越来越多的重视,其作用机制也成为了重要探讨内容。一方面,高糖、高脂等毒性作用会直接对肌小节蛋白产生损伤,另一方面,这些环境因素可能通过增加血流动力学负荷、介导神经内分泌、炎症、能量代谢等机制加重心脏负担,心肌过度收缩,促使心室重构、功能损伤。基于遗传背景,Fumagalli等[48]在研究BMI和HCM风险的相关性时,发现是否携带致病变异并不会影响这种相关性,而Harper等[46]却发现舒张压对发病风险的加重作用在致病变异阴性的人群中更加明显。环境因素平行于致病基因变异发挥作用,抑或是能够直接影响致病基因变异,这一问题仍未得到确切答案,在未来需要结合更多功能验证实验及临床研究来加以解释。
5 治疗进展
现今HCM的药物治疗以 β受体阻滞剂、非二氢吡啶类钙离子通道拮抗剂、丙吡胺、利尿剂类缓解症状为主。近几年来在药物的研究上,减少心肌耗氧代谢类的派克西林、曲美他嗪,通过抑制选择性迟发钠离子通道来降低细胞内钙离子浓度的雷诺嗪都未能有效改善HCM的症状。抑制RAS通路的ARB/ACEI类药物,在病程中晚期的转基因小鼠和HCM患者中无法取得明显疗效,但在心脏形态仍处于正常状态的转基因小鼠中应用能够抑制心肌肥大和纤维化的发生。近期缬沙坦应用于早期HCM患者的二期临床试验表明该药物能够改善患者的心脏结构及功能,但该试验的纳入人群均携带肌小节基因变异,早期使用ARBs的疗效是否存在基因选择性尚未可知[49]。此外,对携带/、/、/的HCM患者使用ACEI类药物治疗,延缓心室肥厚的效果不同,/>/>/。根据以上研究,ARB/ACEI类药物在HCM的治疗上具有前景,但疗效可能受病程长短及基因型的影响。
HCM中大多数致病变异通过增强肌球蛋白ATP酶活性、加快张力增长速度、增多横桥形成、增快滑行速度等机制,导致肌动蛋白–肌球蛋白交互作用加强,引起心肌过度收缩和不全舒张。近期对肌球蛋白构象进行研究发现,HCM的肌球蛋白构象比例失衡,即处于超松弛构象比例增大,无序构象比例减少[50]。以此发病机制作为治疗靶点,最新研发出的Mavacamten可降低肌球蛋白重链上的ATP酶活性,可逆地抑制心肌肌球蛋白和肌动蛋白结合,从而抑制心肌的过度收缩。动物研究表明口服Mavacamten治疗可以抑制甚至逆转心肌肥厚,在疾病早期(心肌过度肥厚出现前)开始治疗还能抑制心肌纤维化的发生。临床上,Mavacamten已完成了用于梗阻性HCM的三期临床试验,在试验中有效改善了梗阻性HCM患者的临床症状、心脏结构功能和生活质量。其中部分患者接受了基因检测,携带致病/可能致病基因变异和意义不明变异的患者中达到主要终点(pVO2和NYHA分级得到显著改善)的占比高于致病变异阴性的患者[51~53],说明该药或许更能改善携带致病变异的患者的症状,因此,药物的疗效是否受到基因型的影响也是未来广泛应用于临床前必不可少的议题。此外,Mavacamten的长期疗效、安全性及和其他治疗方式的对照研究也正在开展(NCT03723655、NCT04349072)。
对于药物难以控制的具有明显症状的梗阻性HCM患者,室间隔外科切除术(septal myectomy,SM)和室间隔酒精消融术(alcohol septal ablation,ASA)是主要的侵入性治疗方式,两者疗效与安全性的影响因素包括术前室间隔厚度及压差、传导束支结构、医疗中心及术者水平等,ASA的疗效与安全性还和间隔支解剖结构及注射酒精量联系密切。关于遗传背景对术式的影响,现有研究表明携带致病基因变异与否在SM和ASA的疗效及安全性方面均未造成显著差异[54,55]。Liwen术式是在超声引导下,经皮肤、肋间、心外膜、心尖心肌内精准穿刺直接送达室间隔肥厚部位并进行射频消融的新兴微创术式,不依赖血管解剖结构,且利于避免损伤传导束支,在中短期内能够有效改善流出道梗阻和症状[56]。今后,该术式的疗效和安全性需要更长期的研究去证明,和其他术式之间的随机对照研究亟需展开。同时,以上研究结果是否受遗传因素影响,也会成为未来必不可少的探讨内容。
除了药物和侵入性方式,基因治疗在遗传病的治疗手段中逐渐崭露头角。在过去的十年里,基因编辑、基因替换、外显子跳跃、等位基因特异性沉默、RNA反式剪接等技术在HCM动物模型或是人类诱导多潜能干细胞实验中展现了治疗潜力,但受限于不足的修复效率、脱靶现象等问题,这些治疗方法到临床应用仍有距离。不久前我国兰峰团队独立研发了具有扩展PAM区的ABEmax-NG系统,使用碱基编辑技术纠正了HCM小鼠胚胎中的致病性()突变,消除了HCM表型[57]。该项新技术在不引入基因插入或缺失的情况下显示出高于70%的修复率,安全性和有效性都得到了验证,展现出了广阔的临床应用前景。
国内外学者通过不懈努力,在各大治疗手段领域中突破壁垒、接连取得重大创新成果,为HCM患者的预后改善带来曙光。
6 结语与展望
随着科技的进步,人们对于HCM的了解逐渐深入,包括更多致病基因、遗传模式被发现。HCM可能不全是一种单基因遗传病,其发病及病程发展受致病基因变异、修饰基因、环境因素等多种因素共同影响,导致了它的表型异质性,探索基因型–表型关联及其他因素对疾病的影响益于更加全面深刻地认识这种疾病,把握疾病自然史,从而施加合理及时的治疗干预来改善预后。但目前为止,基因型–表型联系尚未得到确切答案,遗传背景在HCM的诊治过程中只能起到参考作用,还需大量研究来实现基因指导临床决策。在治疗领域,人们不断创新术式,根据疾病相关基因和机制来研发新型药物及基因治疗手段,使得在该项领域的步伐持续向前迈进。在初步确认疗效和安全性的基础上,遗传背景是否会影响治疗效果将是未来药物试验的重要课题之一。相信今后HCM的遗传背景能够为HCM的诊治提供更多思路与价值。
[1] 中华医学会心血管病学分会中国成人肥厚型心肌病诊断与治疗指南编写组, 中华心血管病杂志编辑委员会. 中国成人肥厚型心肌病诊断与治疗指南. 中华心血管病杂志, 2017, 45(12): 1015–1032.
[2] Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, Kimmelstiel C, Kittleson M, Link MS, Maron MS, Martinez MW, Miyake CY, Schaff HV, Semsarian C, Sorajja P. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines., 2020, 76(25): e159–e240.
[3] Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Consortium EA, MacArthur DG, Farrall M, Cook SA, Watkins H. Reassessment of Mendelian gene pathogenicity using 7, 855 cardiomyopathy cases and 60, 706 reference samples., 2017, 19(2): 192–203.
[4] Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, Cox SW, DePalma SR, Ho CY, Seidman JG, Seidman CE, Rehm HL. Results of clinical genetic testing of 2, 912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity., 2015, 17(11): 880–888.
[5] Ochoa JP, Sabater-Molina M, Garcia-Pinilla JM, Mogensen J, Restrepo-Córdoba A, Palomino-Doza J, Villacorta E, Martinez-Moreno M, Ramos-Maqueda J, Zorio E, Peña-Peña ML, García-Granja PE, Rodríguez-Palomares JF, Cárdenas-Reyes IJ, de la Torre-Carpente MM, Bautista- Pavés A, Akhtar MM, Cicerchia MN, Bilbao-Quesada R, Mogollón-Jimenez MV, Salazar-Mendiguchía J, Mesa Latorre JM, Arnaez B, Olavarri-Miguel I, Fuentes-Cañamero ME, Lamounier Jr A, , Pérez Ruiz JM, Climent-Payá V, Pérez-Sanchez I, Trujillo-Quintero JP, Lopes LR, Repáraz-Andrade A, Marín-Iglesias R, Rodriguez-Vilela A, Sandín-Fuentes M, Garrote JA, Cortel-Fuster A, Lopez-Garrido M, Fontalba-Romero A, Ripoll-Vera T, Llano-Rivas I, Fernandez-Fernandez X, Isidoro-García M, Garcia- Giustiniani D, Barriales-Villa R, Ortiz-Genga M, García- Pavía P, Elliott PM, Gimeno JR, Monserrat L. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy., 2018, 72(20): 2457–2467.
[6] Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C, León D, Martín M, Puente XS, López-Otín C. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy., 2014, 5: 5326.
[7] Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly., 2002, 105(4): 446–451.
[8] Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene., 1999, 262(2): 411–417.
[9] Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis., 2010, 55(11): 1127–1135.
[10] Geier C, Perrot A, Ozcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PF, Fürst DO, Vornwald A, von Hodenberg E, Nürnberg P, Scheffold T, Dietz R, Osterziel KJ. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy., 2003, 107(10): 1390–1395.
[11] Vasile VC, Ommen SR, Edwards WD, Ackerman MJ. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy., 2006, 345(3): 998–1003.
[12] Friedrich FW, Wilding BR, Reischmann S, Crocini C, Lang P, Charron P, Müller OJ, McGrath MJ, Vollert I, Hansen A, Linke WA, Hengstenberg C, Bonne G, Morner S, Wichter T, Madeira H, Arbustini E, Eschenhagen T, Mitchell CA, Isnard R, Carrier L. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy., 2012, 21(14): 3237–3254.
[13] Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy., 2007, 100(6): 766–768.
[14] Eberly LA, Day SM, Ashley EA, Jacoby DL, Jefferies JL, Colan SD, Rossano JW, Semsarian C, Pereira AC, Olivotto I, Ingles J, Seidman CE, Channaoui N, Cirino AL, Han L, Ho CY, Lakdawala NK. Association of race with disease expression and clinical outcomes among patients with hypertrophic cardiomyopathy., 2020, 5(1): 83–91.
[15] Wu GX, Liu LW, Zhou ZY, Liu J, Wang B, Ruan JY, Yang QL, Kanchwala M, Dai PG, Zhang CN, Wang D, Kang LM, Wang S, Y Hui RT, Zou YB, Xing C, Song L, Wang JZ. East Asian-specific common variant in TNNI3 predisposes to hypertrophic cardiomyopathy., 2020, 142(21): 2086–2089.
[16] Vermeer AMC, Clur SB, Blom NA, Wilde AAM, Christiaans I. Penetrance of hypertrophic cardiomyopathy in children who are mutation positive., 2017, 188: 91–95.
[17] Maurizi N, Michels M, Rowin EJ, Semsarian C, Girolami F, Tomberli B, Cecchi F, Maron MS, Olivotto I, Maron BJ. Clinical course and significance of hypertrophic cardiomyopathy without left ventricular hypertrophy., 2019, 139(6): 830–833.
[18] Jensen MK, Havndrup O, Christiansen M, Andersen PS, Diness B, Axelsson A, Skovby F, Køber L, Bundgaard H. Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing., 2013, 127(1): 48–54.
[19] Captur G, Moon JC. Evolution of hypertrophic cardiomyopathy in sarcomere mutation carriers., 2016, 102(22): 1779–1781.
[20] van Velzen HG, Schinkel AFL, Baart SJ, Oldenburg RA, Frohn-Mulder IME, van Slegtenhorst MA, Michels M. Outcomes of contemporary family screening in hypertrophic cardiomyopathy., 2018, 11(4): e001896.
[21] Lorenzini M, Norrish G, Field E, Ochoa JP, Cicerchia M, Akhtar MM, Syrris P, Lopes LR, Kaski JP, Elliott PM. Penetrance of hypertrophic cardiomyopathy in sarcomere protein mutation carriers., 2020, 76(5): 550–559.
[22] Cui H, Schaff HV, Lentz Carvalho J, Nishimura RA, Geske JB, Dearani JA, Lahr BD, Lee AT, Bos JM, Ackerman MJ, Ommen SR, Maleszewski JJ. Myocardial histopathology in patients with obstructive hypertrophic cardiomyopathy., 2021, 77(17): 2159–2170.
[23] Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, Lai A, Amr A, Haas J, Proctor T, Ehlermann P, Jensen K, Katus HA, Meder B. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals., 2018, 107(1): 30–41.
[24] Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe)., 2018, 138(14): 1387–1398.
[25] Lafreniere-Roula M, Bolkier Y, Zahavich L, Mathew J, George K, Wilson J, Stephenson EA, Benson LN, Manlhiot C, Mital S. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines?, 2019, 40(45): 3672–3681.
[26] Lee SP, Ashley EA, Homburger J, Caleshu C, Green EM, Jacoby D, Colan SD, Arteaga-Fernández E, Day SM, Girolami F, Olivotto I, Michels M, Ho CY, Perez MV, Investigators SH. Incident atrial fibrillation is associated with MYH7 sarcomeric gene variation in hypertrophic cardiomyopathy., 2018, 11(9): e005191.
[27] Velicki L, Jakovljevic DG, Preveden A, Golubovic M, Bjelobrk M, Ilic A, Stojsic S, Barlocco F, Tafelmeier M, Okwose N, Tesic M, Brennan P, Popovic D, Ristic A, MacGowan GA, Filipovic N, Maier LS, Olivotto I. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy., 2020, 20(1): 516.
[28] De Bortoli M, Vio R, Basso C, Calore M, Minervini G, Angelini A, Melacini P, Vitiello L, Vazza G, Thiene G, Tosatto S, Corrado D, Iliceto S, Rampazzo A, Calore C. Novel missense variant in MYL2 gene associated with hypertrophic cardiomyopathy showing high incidence of restrictive physiology., 2020, 13(2): e002824.
[29] Liu W, Wei ZK, Zhang YF, Liu Y, Bai RC, Ma CY, Yang J, Sun DD. Identification of three novel pathogenic mutations in sarcomere genes associated with familial hypertrophic cardiomyopathy based on multi-omics study., 2021, 520: 43–52.
[30] Tran Vu MT, Nguyen TV, Huynh NV, Nguyen Thai HT, Pham Nguyen V, Ho Huynh TD. Presence of hypertrophic cardiomyopathy related gene mutations and clinical manifestations in Vietnamese patients with hypertrophic cardiomyopathy., 2019, 83(9): 1908–1916.
[31] Lehman SJ, Tal-Grinspan L, Lynn ML, Strom J, Benitez GE, Anderson ME, Tardiff JC. Chronic calmodulin-kinase II activation drives disease progression in mutation- specific hypertrophic cardiomyopathy., 2019, 139(12): 1517–1529.
[32] Janin A, Bessière F, Chauveau S, Chevalier P, Millat G. First identification of homozygous truncating CSRP3 variants in two unrelated cases with hypertrophic cardiomyopathy., 2018, 676: 110–116.
[33] Salazar-Mendiguchía J, Barriales-Villa R, Lopes LR, Ochoa JP, Rodríguez-Vilela A, Palomino-Doza J, Larrañaga-Moreira JM, Cicerchia M, Cárdenas-Reyes I, García-Giustiniani D, Brögger N, Fernández G, García S, Santiago L, Vélez P, Ortiz-Genga M, Elliott PM, Monserrat L. The p. (Cys150Tyr) variant in CSRP3 is associated with late-onset hypertrophic cardiomyopathy in heterozygous individuals., 2020, 63(12): 104079.
[34] Landstrom AP, Ackerman MJ. Beyond the cardiac myofilament: hypertrophic cardiomyopathy- associated mutations in genes that encode calcium-handling proteins., 2012, 12(5): 507–518.
[35] Wang JZ, Wang YL, Zou YB, Sun K, Wang ZM, Ding H, Yuan JQ, Wei W, Hou Q, Wang H, Liu X, Zhang HJ, Ji Y, Zhou XL, Sharma RK, Wang DW, Ahmad F, Hui RT, Song L. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy., 2014, 16(9): 950–957.
[36] Yuan Y, Meng L, Zhou Y, Lu N. Genetic polymorphism of angiotensin-converting enzyme and hypertrophic cardiomyopathy risk: a systematic review and meta- analysis., 2017, 96(48): e8639.
[37] Zhen Z, Gao L, Wang Q, Chen X, Na J, Xu XW, Yuan Y. Angiotensinogen M235T polymorphism and susceptibility to hypertrophic cardiomyopathy in Asian population: a meta analysis., 2020, 21(4): 1470320320978100.
[38] Rani B, Kumar A, Bahl A, Sharma R, Prasad R, Khullar M. Renin-angiotensin system gene polymorphisms as potential modifiers of hypertrophic and dilated cardiomyopathy phenotypes., 2017, 427(1–2): 1–11.
[39] Wang S, Wang J, Zou Y, Wang J, Wang H, Hui R. Angiotensinogen gene variations and LV outflow obstruction in hypertrophic cardiomyopathy., 2014, 39(2): 258–263.
[40] Mouton JM, van der Merwe L, Goosen A, Revera M, Brink PA, Moolman-Smook JC, Kinnear C. MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients., 2016, 135(5): 477–483.
[41] Zhang C, Zhang HJ, Wu GX, Luo XL, Zhang CN, Zou YB, Wang H, Hui RT, Wang JZ, Song L. Titin-truncating variants increase the risk of cardiovascular death in patients with hypertrophic cardiomyopathy., 2017, 33(10): 1292–1297.
[42] Zhang XL, Xie J, Zhu SH, Chen YH, Wang L, Xu B. Next-generation sequencing identifies pathogenic and modifier mutations in a consanguineous Chinese family with hypertrophic cardiomyopathy., 2017, 96(24): e7010.
[43] Noyes AM, Zhou AY, Gao G, Gu LZ, Day S, Andrew Wasserstrom J, Dudley SC. Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy., 2017, 249: 282–286.
[44] Larsen CM, Ball CA, Hebl VB, Ong KC, Siontis KC, Olson TP, Ackerman MJ, Ommen SR, Allison TG, Geske JB. Effect of body mass index on exercise capacity in patients with hypertrophic cardiomyopathy., 2018, 121(1): 100–106.
[45] Wasserstrum Y, Barriales-Villa R, Fernández-Fernández X, Adler Y, Lotan D, Peled Y, Klempfner R, Kuperstein R, Shlomo N, Sabbag A, Freimark D, Monserrat L, Arad M. The impact of diabetes mellitus on the clinical phenotype of hypertrophic cardiomyopathy., 2019, 40(21): 1671–1677.
[46] Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, Waring A, Ormondroyd E, Kramer CM, Ho CY, Neubauer S, Tadros R, Ware JS, Bezzina CR, Farrall M, Watkins H. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity., 2021, 53(2): 135–142.
[47] Xu HB, Wang J, Yuan JS, Hu FH, Yang WX, Guo C, Luo XL, Liu R, Cui JG, Gao XJ, Chun YS, Qiao SB. Implication of apnea-hypopnea index, a measure of obstructive sleep apnea severity, for atrial fibrillation in patients with hypertrophic cardiomyopathy., 2020, 9(8): e015013.
[48] Fumagalli C, Maurizi N, Day SM, Ashley EA, Michels M, Colan SD, Jacoby D, Marchionni N, Vincent-Tompkins J, Ho CY, Olivotto I, Investigators S. Association of obesity with adverse long-term outcomes in hypertrophic cardiomyopathy., 2020, 5(1): 65–72.
[49] Ho CY, Day SM, Axelsson A, Russell MW, Zahka K, Lever HM, Pereira AC, Colan SD, Margossian R, Murphy AM, Canter C, Bach RG, Wheeler MT, Rossano JW, Owens AT, Bundgaard H, Benson L, Mestroni L, Taylor MRG, Patel AR, Wilmot I, Thrush P, Vargas JD, Soslow JH, Becker JR, Seidman CE, Lakdawala NK, Cirino AL, Investigators V, Burns KM, McMurray JJV, MacRae CA, Solomon SD, Orav EJ, Braunwald E. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial., 2021, 27(10): 1818–1824.
[50] Toepfer CN, Garfinkel AC, Venturini G, Wakimoto H, Repetti G, Alamo L, Sharma A, Agarwal R, Ewoldt JF, Cloonan P, Letendre J, Lun M, Olivotto I, Colan S, Ashley E, Jacoby D, Michels M, Redwood CS, Watkins HC, Day SM, Staples JF, Padrón R, Chopra A, Ho CY, Chen CS, Pereira AC, Seidman JG, Seidman CE. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy., 2020, 141(10): 828–842.
[51] Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE. A small- molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice., 2016, 351(6273): 617–621.
[52] Spertus JA, Fine JT, Elliott P, Ho CY, Olivotto I, Saberi S, Li WY, Dolan C, Reaney M, Sehnert AJ, Jacoby D. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomised, double-blind, placebo- controlled, phase 3 trial., 2021, 397(10293): 2467–2475.
[53] Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, Kubanek M, Wojakowski W, Jensen MK, Gimeno-Blanes J, Afshar K, Myers J, Hegde SM, Solomon SD, Sehnert AJ, Zhang D, Li WY, Bhattacharya M, Edelberg JM, Waldman CB, Lester SJ, Wang A, Ho CY, Jacoby D, investigators E-Hs. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial., 2020, 396(10253): 759–769.
[54] Chauvette V, Accad AJ, Georges G, Bouhout I, Garceau P, L'Allier P, Bouchard D. Septal myectomy in the era of genetic testing., 2021, 36(4): 1282–1288.
[55] Bonaventura J, Norambuena P, Votýpka P, Hnátová H, Adlová R, Macek Jr M, Veselka J. Patients with hypertrophic obstructive cardiomyopathy after alcohol septal ablation have favorable long-term outcome irrespective of their genetic background., 2020, 10(2): 193–200.
[56] Liu LW, Li J, Zuo L, Zhang JZ, Zhou MY, Xu B, Hahn RT, Leon MB, Hsi DH, Ge JB, Zhou XD, Zhang J, Ge SP, Xiong LZ. Percutaneous intramyocardial septal radiofrequency ablation for hypertrophic obstructive cardiomyopathy., 2018, 72(16): 1898–1909.
[57] Ma SH, Jiang WJ, Liu XJ, Lu WJ, Qi T, Wei JJ, Wu FJ, Chang Y, Zhang SY, Song YB, Bai R, Wang JB, Lee AS, Zhang HJ, Wang YM, Lan F. Efficient correction of a hypertrophic cardiomyopathy mutation by ABEmax-NG., 2021, 129(10): 895–908.
Research progress of the correlation between genotype and phenotype in hypertrophic cardiomyopathy
Tian Shu1,2, Haochang Hu1,2, Caijie Shen1, Shaoyi Lin1, Xiaomin Chen1,2
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disease characterized by left ventricular hypertrophywith prevalence of 1/500–1/200. Up to now, 1500 mutations in more than 30 genes have been found to be related to the disease. Pathogenic gene mutations together with polymorphisms of modifying genes and environmental factors play various roles in the disease processes, resulting in phenotypic heterogeneity of the disease, ranging from no symptoms to sudden cardiac death. The pathological phenotypes of HCM mainly include cardiomyocyte hypertrophy, disordered array, fibrosis, myocardial ischemia, and others. In recent years, many research efforts have been devoted to exploring the influence of HCM genotype on phenotype, and development of treatment methods based on genetics. This article focuses on the correction between HCM genotype and phenotype and summarizes the research progresses onHCM in terms of pathogenic genes, pathogenesis, associated modification factors and treatment methods, thereby providing insights on the future research and development on the genetics of HCM.
hypertrophic cardiomyopathy; pathogenic genes; phenotype; treatment
2021-11-07;
2021-12-27;
2022-01-10
舒甜,在读硕士研究生,专业方向:心血管内科。E-mail: shutian08@163.com
陈晓敏,硕士,主任医师,研究方向:心血管内科。E-mail: chxmin@hotmail.com
10.16288/j.yczz.21-324
(责任编委: 张岩)
