V2O5-Al2O3催化CO2氧化乙苯脱氢的稀土氧化物助剂效应
2022-03-18杨国庆宋永红赵永华刘昭铁刘忠文
王 欢,杨国庆,宋永红,赵永华,刘昭铁,刘忠文*
(1 辽宁工业大学 化学与环境工程学院,辽宁 锦州 121001; 2 陕西省合成气转化重点实验室,陕西师范大学 化学化工学院,陕西 西安 710119)
苯乙烯(C8H8,ST)是苯取代乙烯上的1个H原子后形成的一种重要基础有机化工原料。作为仅次于聚乙烯(PE)、聚氯乙烯(PVC)、环氧乙烷(EO)的第四大乙烯衍生物,苯乙烯主要用作树脂、离子交换树脂和合成橡胶的生产原料,且其在合成医药、染料、农药等方面的应用日趋广泛[1]。从苯乙烯下游产品的应用领域来看,随着建材、家电、汽车等与人们生活相关工业的稳步发展,苯乙烯的需求量将有递增趋势[2-3]。目前,苯乙烯主要由乙苯(EB)直接脱氢获得,其产量约占工业苯乙烯总产量的90%,该工艺通常采用Fe-K系氧化物催化剂在过热水蒸气(615~645 ℃)的常压或负压条件下进行[4]。尽管研究人员已对乙苯直接脱氢工艺的活性组分、助剂及参数等方面进行了广泛研究,但依然存在苯乙烯单程收率低及能耗大等问题[5-6]。为了解决上述问题,实现苯乙烯的环保、节能、高效生产,近年来研究人员先后提出了以O2、N2O、CO2作为氧化剂进行乙苯氧化脱氢的策略,其中以CO2为弱氧化剂的乙苯氧化脱氢技术(CO2-ODEB)备受关注[7-9]。
一方面,CO2的弱氧化性能够缓解乙苯直接脱氢的热力学限制,提高乙苯转化率[10-11];另一方面,与O2相比,在CO2作为氧化剂的反应中不存在深度氧化问题,可以显著提高苯乙烯的选择性[9];更为重要的是,该过程绿色环保,有望实现CO2的资源化利用,在提高苯乙烯收率的同时还可以副产CO。然而,由于CO2的参与,CO2-ODEB反应过程与过热蒸气条件下的乙苯直接脱氢工艺并不相同,商业Fe-K系氧化物催化剂不适用于CO2-ODEB反应。近年来,研究人员针对CO2-ODEB催化剂进行了大量筛选和研发,研究对象主要集中在 Fe、Cr、Ce、V、Zr等金属氧化物催化剂上[12-15],但目前报道的催化剂普遍存在快速失活问题,限制了CO2-ODEB绿色工艺的工业化应用。
与其他金属氧化物相比,负载型钒氧化物(VOx)催化剂的活性和稳定性相对较好,且不存在安全和环境问题。通过分析CO2-ODEB的反应特点并结合钒基氧化物催化剂的相关研究进展可以发现,负载型VOx催化CO2-ODEB反应可能遵循Mars-van Krevelen氧化还原机理,VOx的含量、结构、氧化还原能力等是影响其催化性能的关键因素[16-18]。采用合适的制备方法和策略提高钒物种的分散度,同时强化CO2的活化,使之与V5+/V4+的氧化还原循环相匹配,并抑制V5+深度还原为V3+,可能是进一步改善钒基氧化物的CO2-ODEB催化性能尤其是稳定性的重要途径[19]。
研究发现,利用蒸发诱导自组装(EISA)法制备的有序介孔氧化铝(OMA)不仅具有较大的比表面积、孔容以及规整有序的孔道结构,而且能够提高活性组分的分散度,被广泛用作催化剂载体[20-22]。Zhu等采用浸渍法将VOx负载于有序介孔氧化铝上,发现钒与OMA之间存在较强的相互作用,能够显著提高VOx的分散度,因而获得了活性和稳定性都较好的钒基氧化物催化剂[23]。此外,稀土元素具有多种能级结构以及特殊的4f电子层排列方式,很容易得到或失去电子,以Ce为代表的稀土及稀土复合氧化物在氧化和氧化脱氢等化学反应中具有良好的催化或助催化作用[24-25]。目前,稀土氧化物用于CO2-ODEB的研究主要集中于考察某一特定稀土氧化物对钒氧化物结构性能的影响[26-27],或者是将稀土氧化物单独作为活性组分进行研究[14],而关于不同稀土氧化物作为助剂影响负载型VOx催化CO2-ODEB反应的研究却鲜有报道。
基于此,本研究在综合分析负载型VOx催化CO2-ODEB反应和EISA法制备介孔氧化物特点的基础上,拟以VOx为活性组分,Al2O3为载体,采用尿素辅助水热和EISA相结合的方法,制备5种含有不同稀土氧化物(CeO2、Pr6O11、Nd2O3、La2O3、Yb2O3)助剂的钒基复合氧化物催化剂,并考察各催化剂催化CO2-ODEB反应的性能。采用低温N2吸脱附、X射线衍射(XRD)、H2程序升温还原(H2-TPR)、拉曼光谱(Raman)、X射线光电子能谱(XPS)、热分析(TG-DSC)等表征技术系统地研究不同稀土氧化物对钒氧化物催化剂织构及结构特征、还原能力、表面元素价态、积炭行为的影响。通过关联表征与反应评价明确不同稀土氧化物助剂对钒氧化物催化CO2-ODEB反应的作用机制。
1 材料与方法
1.1 催化剂的制备
采用尿素辅助水热和EISA相结合的方法,制备一系列以稀土氧化物(CeO2、Pr6O11、Nd2O3、La2O3、Yb2O3)为助剂、V2O5为活性组分、Al2O3为载体的催化剂。其中,V2O5质量分数为9%,助剂(LnO)质量分数为7%。所制备的催化剂记做V2O5-LnO-Al2O3。
以V2O5-CeO2-Al2O3为例,催化剂的制备过程如下:将0.82 g硝酸铈铵(分析纯,国药集团化学试剂有限公司)和0.18 g尿素(分析纯,国药集团化学试剂有限公司)溶于15 mL去离子水中并转移至水热釜,80 ℃维持6 h,升温至180 ℃维持24 h。样品自然冷却至室温后,将得到的沉淀离心、洗涤,加入到含有22.82 g硝酸铝(分析纯,国药集团化学试剂有限公司)的48 mL乙醇水溶液(乙醇体积分数为80%)中,搅拌至完全溶解,再转移到培养皿中,80 ℃保持5 h。另取7.29 g聚环氧乙烷-聚环氧丙烷-聚环氧乙烷三嵌段共聚物(P123,西格玛奥德里奇贸易有限公司)溶解于121 mL无水乙醇中,先加入2.43 g柠檬酸(分析纯,国药集团化学试剂有限公司)、0.22 g草酸(分析纯,国药集团化学试剂有限公司)和0.43 g偏钒酸铵(分析纯,天津市北辰方正试剂厂),再加入上述培养皿中的样品,室温下剧烈搅拌24 h,然后45 ℃保持48 h。将获得的凝胶在80 ℃烘箱中干燥10 h,550 ℃马弗炉中煅烧4 h,得到V2O5-CeO2-Al2O3催化剂。
采用同样方法,通过改变前驱体的种类和用量,分别制备V2O5-Pr6O11-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3和V2O5-Yb2O3-Al2O3催化剂。
1.2 催化剂的性能评价及产物分析
催化剂性能测试在内径为10 mm的不锈钢微型固定床反应管中进行。常压下,将0.5 g(40~60 目)催化剂样品与等量、等目数的石英砂均匀混合,置于反应管的恒温区。在20 mL/min 的N2气氛下,将反应床层温度以10 ℃/min的速率升温至550 ℃,稳定1 h后切换成CO2气氛(20 mL/min)保持15 min。用微量泵以0.006 mL/min的速率打入乙苯,乙苯经气化器气化并与CO2充分混合后进入反应器,其中,n(CO2)∶n(EB)=20,接触时间为0.17 h。反应产生的苯、甲苯、苯乙烯等产物以及未反应的乙苯经冷却收集后,使用SP-6800A气相色谱仪(山东鲁南瑞虹化工仪器有限公司)进行定性与定量分析。仪器配置FID检测器和HP-5(0.25 mm×30 m)毛细柱,分析条件为:进样口温度160 ℃,柱温150 ℃,检测器温度180℃,进样量0.2 μL。定量分析采用峰面积归一法。
1.3 催化剂的表征
1.3.1 N2吸脱附
使用物理吸附仪(BelSorp-Max,日本BEL公司)对催化剂的织构和结构特征进行测试。样品首先在300 ℃、真空条件下脱气10 h,然后在-196 ℃条件下进行N2吸脱附测试。通过N2吸附量计算催化剂的孔体积,采用BET方程计算催化剂的比表面积,根据BJH模型得到催化剂的孔径分布曲线。
1.3.2 X射线衍射(XRD)
采用X-射线粉末衍射仪(D8 Advance,德国Bruker公司)对催化剂的结构和物相进行测试。以Cu靶 Kα为射线源,管电压为40 kV,管电流为40 mA,扫描速率为5(°)/min,小角XRD测试范围为0.5°~10°,广角XRD测试范围为10°~85°。
1.3.3 氢气程序升温还原(H2-TPR)
使用化学吸附仪(Autochem 2920,美国麦克仪器公司)测定催化剂的还原性能。取40~60目催化剂50 mg装入U型石英管中,Ar气氛下以10 ℃/min的速率升温至300 ℃,保持1 h,去除催化剂表面吸附的水和杂质;然后降温至50 ℃,切换为30 mL/min的H2/Ar混合气(H2体积分数为10%),待TCD信号稳定后以10 ℃/min的速率程序升温至1 000 ℃。通过TCD记录耗氢信号。
1.3.4 拉曼光谱(Raman spectra)
使用显微共聚焦激光拉曼光谱仪(Via Reflex Raman,英国雷尼绍公司)对催化剂的结构进行表征。选用325 nm的紫外光在室温环境下对催化剂样品进行检测,测量范围为200~1 400 cm-1。
1.3.5 X射线光电子能谱(XPS)
催化剂反应前后的元素价态测定在X射线光电子能谱仪(AXIS ULTRA DLD,日本Kratos Analytical Ltd.公司)上进行。以单色器的铝靶为X射线源(1 486.6 eV),在室温、高真空条件下进行测定,以催化剂样品表面污染碳C 1s(结合能为284.8 eV)为基准对电子结合能进行校定。
1.3.6 热分析(TG-DSC)
催化剂反应后的积炭测试在热重-差热分析仪(STA449 F5,德国Netzsch公司)上进行。取10 mg左右催化剂样品置于坩埚中,在100 mL/min的空气气氛下以10 ℃/min的速率从室温程序升温至1 000 ℃,测定样品的热重和差热曲线。
2 结果与讨论
2.1 催化剂的织构及结构特征
所制备催化剂的N2吸脱附曲线及孔径分布曲线如图1所示。从N2吸脱附曲线可以看出(图1a),当相对压力小于0.1时,制备的催化剂都表现出明显的氮气吸附,说明催化剂上存在少量微孔。当相对压力增加至0.5~0.8时,5种催化剂曲线都出现了明显的具有H1型滞后环的Ⅳ型吸附等温线,说明5种催化剂都以均匀的圆柱形介孔结构为主[20]。由图1b的孔径分布结果可以看出,所有催化剂的孔径分布范围都较窄,主要集中在4~12 nm之间,进一步说明催化剂存在较为均匀的介孔结构。
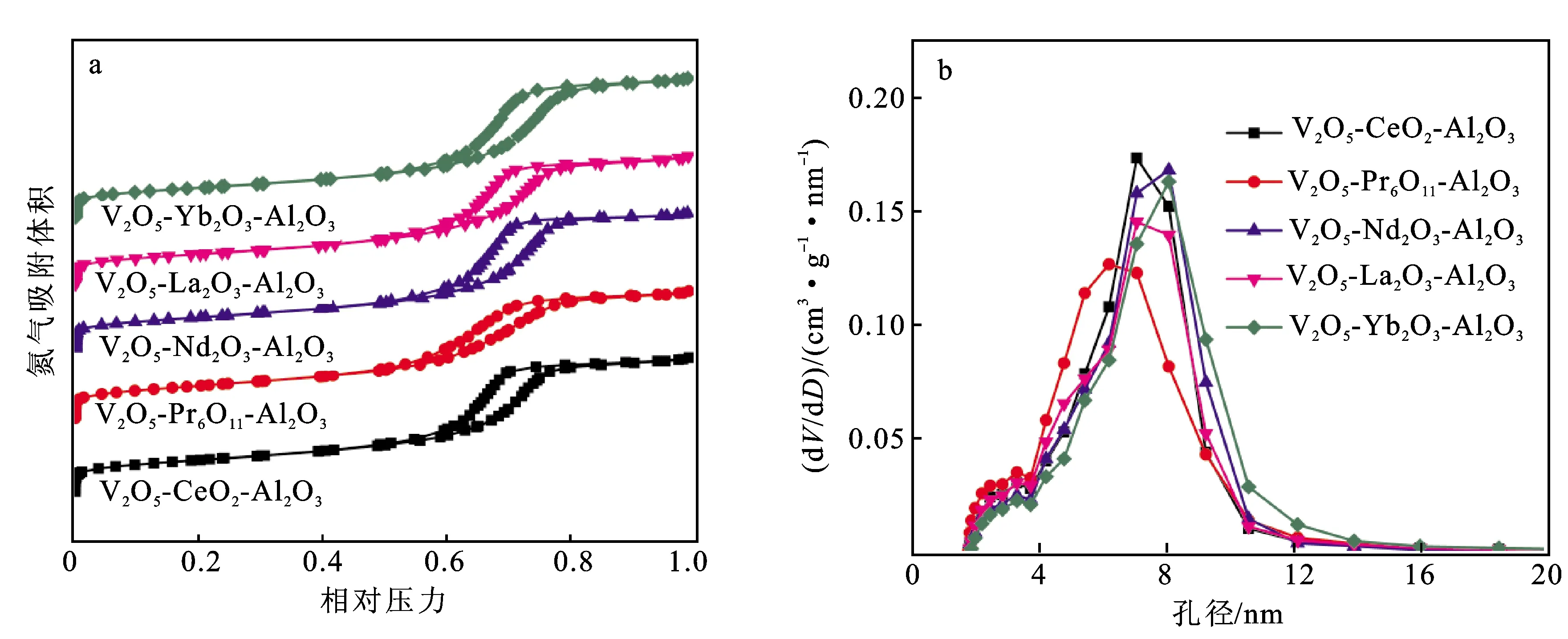
V代表孔体积,D代表孔径。图1 不同催化剂的N2吸脱附曲线(a)及孔径分布曲线(b)Fig.1 N2 adsorption-desorption isotherms (a) and pore-size distribution curves (b) of different catalysts
表1展示了5种催化剂的比表面积和孔结构参数。可以看出,通过尿素辅助水热和EISA相结合方法制备的5种钒基复合氧化物催化剂具有相似的织构性质,比表面积较高,在357~393 m2/g之间。各催化剂的比表面积由大到小依次为:V2O5-Pr6O11-Al2O3、V2O5-CeO2-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3、V2O5-Yb2O3-Al2O3,而孔径和孔体积则按照相反的顺序排列。值得注意的是,V2O5-Pr6O11-Al2O3催化剂的平均孔径和孔体积最小,比表面积最大。
图2a为所制备催化剂的小角XRD图。由图可知,所有催化剂均在1.2°和2.0°附近出现有一强一弱2个峰,分别归属于p6mm六方相(100)晶面和(110)晶面的特征衍射峰,说明所制备催化剂具有规整有序的介孔结构[18,20]。 此外, 添加稀土氧化物的种类不同,催化剂(100)晶面衍射峰的强度也不同,说明不同稀土氧化物助剂对催化剂孔道结构的有序度或规整性具有不同程度的影响,其中V2O5-CeO2-Al2O3催化剂的孔道结构有序度或规整性最好,而V2O5-Yb2O3-Al2O3催化剂相对较差。

表1 不同催化剂的比表面积及孔结构参数Tab.1 Specific surface area and pore structure parameters of different catalysts
所制备催化剂的广角XRD图如图2b所示。V2O5-Pr6O11-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3、V2O5-Yb2O3-Al2O3催化剂仅在26°和66°附近检测到2个弥散的馒头峰,表明在这4种催化剂中氧化铝载体、钒氧化物和添加的稀土氧化物助剂以高分散或无定型状态存在。相反,V2O5-CeO2-Al2O3催化剂在28.5°、33.1°、47.5°、56.5°、69.6°、77.0°和79.2°处均检测到明显的归属于立方相CeO2的特征峰(PDF#34-0394)。综上,N2吸脱附和XRD表征结果均表明不同稀土氧化物助剂对钒基氧化物催化剂的织构及结构特征影响不同。

图2 不同催化剂的小角XRD(a)和广角XRD(b)图Fig.2 Small-angle XRD(a) and wide-angle XRD(b) patterns of different catalysts
为进一步探明所制备催化剂中钒氧化物的结构信息,对5种催化剂进行了紫外拉曼光谱表征,结果如图3所示。可以看出,5种催化剂都在486、945和1 015 cm-1(肩峰)附近都出现了分别归属于V-O-V、V-O-Al和V=O的拉曼特征峰[17],同时催化剂样品中都未检测到归属于结晶态V2O5的拉曼特征峰(995 cm-1),表明5种催化剂中的钒结构相似且分散度较高,全部以孤立态和聚合态共存的形式分散于载体表面。这可能是由于通过尿素辅助水热和EISA相结合方法制备的钒基氧化物催化剂具有较高的比表面积(表1),且催化剂中规整的介孔氧化铝和钒氧化物之间存在较强的相互作用[23],钒物种能够较好地分散在载体表面。

图3 不同催化剂的紫外拉曼光谱Fig.3 UV Raman spectra of different catalysts
2.2 催化剂的还原能力及表面元素
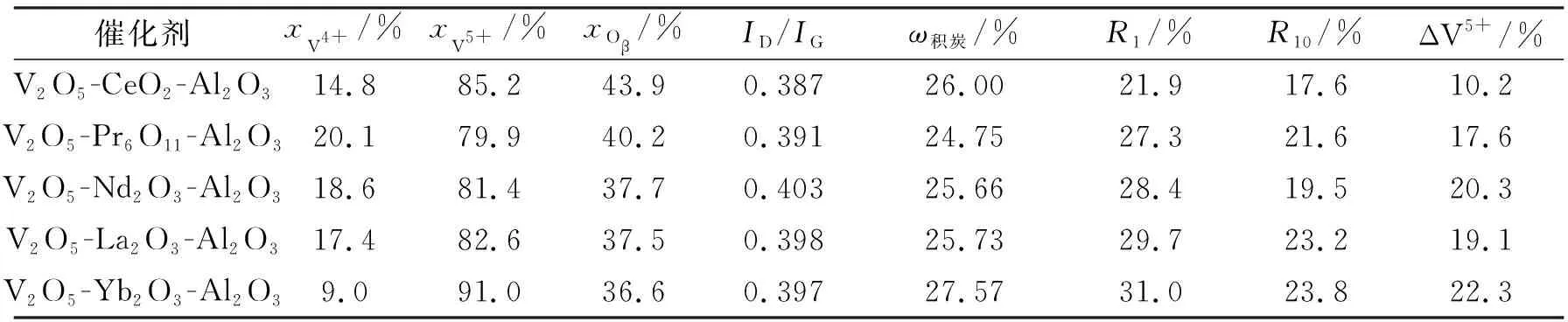
采用H2-TPR探究不同稀土氧化物掺杂对钒基催化剂还原能力的影响,结果如图4所示。所制备的5种催化剂都在500 ℃和770 ℃附近出现了2个明显的还原峰,其中,前者归属于载体Al2O3表面高度分散的钒物种的还原,后者归属于少量体相钒物种从V5+到V3+的还原[18]。值得注意的是,催化剂中所加入稀土氧化物的种类不同,表面钒物种的最大还原峰温度(500 ℃附近)也不同。各催化剂的最大还原峰温度由低到高依次为:V2O5-Nd2O3-Al2O3 图4 不同催化剂的H2-TPR图谱Fig.4 H2-TPR profiles of different catalysts 为了分析催化剂表面钒氧化物的价态和氧物种,对所制备催化剂进行了XPS表征。图5a为不同催化剂V 2p3/2的XPS谱图。所有催化剂在517 eV附近都检测到1个较宽且不对称的V 2p3/2特征峰,说明钒物种中存在多种价态。其中,V2O5-CeO2-Al2O3的V 2p3/2特征峰结合能为517.2 eV,略低于其他4种催化剂的特征峰结合能(517.4~517.5 eV)。一般来说,V 2p3/2结合能除了与价态有关外,一定程度上还受到钒物种与载体或助剂之间相互作用的影响。考虑到V2O5-CeO2-Al2O3中的CeO2为结晶态(图2),而其他催化剂中的稀土氧化物助剂都以高分散或无定型形式存在,V2O5-CeO2-Al2O3的V 2p3/2特征峰向低结合能偏移可能是因为结晶态的CeO2使得V2O5-CeO2-Al2O3中钒氧化物的配位结构与其他催化剂不同。 进一步将V 2p3/2峰进行高斯拟合[28],发现样品中的钒物种主要以517.6 eV处的V5+为主,同时也存在少量V4+(516.6 eV附近)。为了进行定量比较,利用峰面积计算V4+和V5+的相对物质的量分数,结果列于表2。加入不同稀土氧化物后,各催化剂表面V5+物种的相对物质的量分数不同,由小到大依次为:V2O5-Pr6O11-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3、V2O5-CeO2-Al2O3、V2O5-Yb2O3-Al2O3。值得注意的是,V2O5-Yb2O3-Al2O3催化剂中V5+的相对物质的量分数最高(91.0%),但其V 2p3/2峰面积最小,说明该催化剂中表面钒物种的数量最少,这与H2-TPR结果中其具有最小表面钒物种还原峰的结果是一致的(图4)。 图5b给出了不同催化剂O 1s的XPS谱图。可以看出,所有催化剂在528~535 eV之间均具有1个非常宽的O 1s谱峰。为了区别氧物种的类型,采用高斯-洛伦兹模型将O 1s谱图拟合成位于530.3~530.6 eV、531.4~531.8 eV和532.5~533.1 eV的离子化峰,分别归属于表面晶格氧(Oα)、表面吸附氧(Oβ)、化学吸附水和/或碳酸盐中的氧(Oγ)[14]。其中,由表面氧缺陷带来的具有不饱和化学键的Oβ被认为是化学反应中的活性氧物种,在氧化反应或氧化脱氢反应中发挥着关键作用。因此,根据各个氧物种的峰面积计算了Oβ的相对物质的量分数(表2),其中,V2O5-CeO2-Al2O3催化剂中的Oβ相对物质的量分数最高(43.9%),V2O5-Pr6O11-Al2O3次之(40.2%),其余3个催化剂中的Oβ相对物质的量分数差异不大,均在37%左右。以上结果表明,不同种类的稀土氧化物掺杂能够显著影响催化剂表面的钒物种价态及氧缺陷含量。 图5 不同催化剂的V 2p3/2(a)和O 1s(b)XPS谱图Fig.5 V 2p3/2 (a) and O 1s (b) XPS spectra of different catalysts 表2 不同催化剂的XPS数据拟合结果及反应后数据汇总Tab.2 The peak-fitting results of XPS spectra and the summarized reaction results of different catalysts 5种所制备催化剂的CO2-ODEB反应性能如图6所示。反应稳定后,所有催化剂的苯乙烯选择性都在98%左右(图6a),其表现出的较高选择性与文献报道的钒氧化物催化剂的选择性结果一致[19]。由图6b中的催化活性结果可以看出,不同催化剂的乙苯转化率差异较大。其中,V2O5-Pr6O11-Al2O3催化剂的初始活性最高,其乙苯初始转化率(反应时间1 h)为64.9%;而V2O5-Yb2O3-Al2O3催化剂的初始活性最低,其乙苯初始转化率仅为57.7%。各催化剂的乙苯初始转化率由大到小依次为:V2O5-Pr6O11-Al2O3>V2O5-CeO2-Al2O3>V2O5-La2O3-Al2O3≈V2O5-Nd2O3-Al2O3>V2O5-Yb2O3-Al2O3。随着反应时间的延长,所有催化剂的乙苯转化率都有所下降,反应50 h时5种催化剂的乙苯转化率由大到小依次为:V2O5-CeO2-Al2O3>V2O5-Pr6O11-Al2O3>V2O5-Nd2O3-Al2O3≈V2O5-La2O3-Al2O3>V2O5-Yb2O3-Al2O3。 为了进一步量化催化剂的失活程度,分别计算出催化剂的相对失活速率R1和R10。其中,R1=(X1-X50)/X1×100%,R10=(X10-X50)/X1×100%,X1、X10和X50分别为反应1 h、10 h和50 h时的乙苯转化率。结果表明(表2),V2O5-CeO2-Al2O3催化剂的R1为21.9%,R10为17.6%,明显低于其他催化剂的R1和R10,说明其稳定性最好。 以上结果表明,不同种类的稀土氧化物助剂会显著影响钒基催化剂的活性和稳定性。V2O5-Pr6O11-Al2O3催化剂具有最高初始活性(64.9%),V2O5-CeO2-Al2O3催化剂的初始活性(62.9%)略低于V2O5-Pr6O11-Al2O3催化剂,但随着反应时间的延长,其稳定性最好。与已报道的钒基催化剂上CO2-ODEB反应活性的结果相比[18],采用尿素辅助水热和EISA相结合方法制备的含稀土氧化物助剂的钒基催化剂表现出更高的催化反应活性和长周期稳定性。 图6 不同催化剂的苯乙烯选择性(a)和乙苯转化率(b)Fig.6 The styrene selectivities (a) and ethylbenzene conversion rates (b) of different catalysts 通过关联催化剂的表征和性能评价结果,并结合CO2-ODEB反应可能的Mars-van Krevelen氧化还原机理,可以看出不同稀土氧化物助剂对钒基氧化物催化剂的织构特性、结构特征及氧化还原性能的影响是不同的,进而显著影响了催化剂的活性和稳定性。所制备的催化剂中,V2O5-Pr6O11-Al2O3催化剂的比表面积最大,孔道规整度较高,可还原的表面钒物种最多,表现出最高的乙苯初始转化率。相反,V2O5-Yb2O3-Al2O3催化剂的比表面积最小,孔道规整度最差,可还原的表面钒物种数量最少,表现出最低的反应活性。值得注意的是,V2O5-CeO2-Al2O3催化剂的比表面积适中,孔道结构规整,还原温度适宜,可还原的表面钒物种较多,氧缺陷含量最高,不仅表现出仅次于V2O5-Pr6O11-Al2O3催化剂的较高初始活性,而且具有最佳稳定性。基于上述认识,为了进一步确定稀土氧化物对催化剂活性及稳定性的关键影响因素,本研究对反应后催化剂进行了Raman、TG-DSC和XPS表征。 图7为反应50 h时催化剂在空气气氛下的TG-DSC曲线。反应后5种催化剂的TG曲线在380~520 ℃之间都存在1个明显的失重峰(图7a),且在相同温度区间对应的DSC曲线中都可以观察到1个明显的放热峰(图7b),这是由反应后催化剂表面沉积的焦炭与空气中的氧气发生燃烧反应造成的[18]。此外,比较图7b中不同催化剂的DSC曲线可以发现,5种催化剂由积炭燃烧而产生的放热峰形状基本相同,放热峰位置也比较接近(469.0±5.0 ℃),说明不同催化剂在反应过程中形成的积炭物种相似。 进一步采用可见拉曼对反应后催化剂的积炭物种进行鉴定,结果如图8所示。所有催化剂的Raman图谱都在1 360 cm-1和1 600 cm-1处出现了特征峰,分别代表无定型炭的D键和石墨型炭的G键[14, 18]。为了量化催化剂表面积炭的石墨化程度,计算了D键和G键的强度比(ID/IG),发现各催化剂的ID/IG值都在0.395±0.008之间(表2),说明各催化剂积炭的石墨化程度相似,这与上述TG-DSC的表征结果一致。 图7 反应50 h时不同催化剂的TG曲线(a)和DSC曲线(b)Fig.7 TG (a) and DSC (b) profiles of different catalysts after CO2-ODEB of 50 h 一般来说,反应过程中催化剂表面的积炭行为是催化剂失活的原因之一。为了明确积炭行为与催化剂失活之间的关系,根据图7a结果计算了各催化剂的积炭量(表2)。不同催化剂反应后的积炭量从小到大依次为:V2O5-Pr6O11-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3、V2O5-CeO2-Al2O3、V2O5-Yb2O3-Al2O3。根据相关文献[16, 29]报道,催化剂积炭量与乙苯转化率密切相关,催化剂累计转化的乙苯越多,其表面产生的积炭也越多。但在本研究中,V2O5-Pr6O11-Al2O3具有最高乙苯转化率,积炭量却最少;而V2O5-Yb2O3-Al2O3活性最差,转化的乙苯量最少,积炭量却最多。这说明Pr6O11掺杂对催化剂积炭具有较好的抑制作用,而Yb2O3的抗积炭能力较差。此外,催化剂的失活速率按如下顺序递增:V2O5-CeO2-Al2O3、V2O5-Pr6O11-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-La2O3-Al2O3、V2O5-Yb2O3-Al2O3(表2)。对比以上结果可以明显看出,积炭量和相对失活速率,即催化剂的稳定性,并不是一一对应关系。相反,在所制备的催化剂中,V2O5-CeO2-Al2O3的积炭量虽然仅次于V2O5-Yb2O3-Al2O3,却表现出最佳稳定性,说明积炭可能不是导致本研究中催化剂失活的关键原因,这与前期的研究结果一致[14, 18]。 图8 反应50 h时不同催化剂的拉曼谱图Fig.8 Raman spectra of different catalysts after CO2-ODEB of 50 h 结合CO2-ODEB可能的Mars-van Krevelen氧化还原机理[17, 19],推测反应过程中V5+的不可逆还原可能是导致催化剂逐渐失活的关键因素。基于此,本研究对反应50 h时的催化剂进行了XPS表征,并计算了反应前后各催化剂表面V5+的相对物质的量分数差ΔV5+(表2)。反应前后各催化剂的ΔV5+值按如下顺序递增:V2O5-CeO2-Al2O3、V2O5-Pr6O11-Al2O3、V2O5-La2O3-Al2O3、V2O5-Nd2O3-Al2O3、V2O5-Yb2O3-Al2O3,说明V2O5-CeO2-Al2O3催化剂在催化CO2-ODEB反应的过程中能够很好地抑制V5+的不可逆还原,进而最大程度地维持V5+物种的存在,这可能是其具有最佳稳定性的关键原因。此外,结合各催化剂反应前的Oβ相对物质的量分数(表2)可知,添加CeO2后催化剂中的氧缺陷含量最多,说明V2O5-CeO2-Al2O3催化剂能够有效活化CO2,补充消耗的晶格氧[14]。根据氧化还原机理可知,晶格氧能够将被还原的V4+再次氧化为V5+,从而维持V5+与V4+之间的氧化还原循环稳定性,因此V2O5-CeO2-Al2O3催化剂表现出最佳的长周期稳定性。在所有制备的催化剂中,V2O5-Yb2O3-Al2O3催化剂表面的可还原钒物种和氧缺陷数量最少,因此活性和稳定性都最差。 本研究将尿素辅助水热法和蒸发诱导自组装法相结合,制备了一系列以Al2O3为载体、以不同稀土氧化物(CeO2、Pr6O11、Nd2O3、La2O3、Yb2O3)为助剂的钒基复合氧化物催化剂,并考察了不同稀土氧化物对钒基氧化物催化CO2-ODEB反应性能的影响。所制备的催化剂不仅具有较大的比表面积和较为规整的孔道结构,而且其钒氧化物都以孤立和聚集态共存的形式高度分散在Al2O3表面。不同稀土氧化物助剂对催化剂的结构、化学性质和CO2-ODEB反应性能影响不同。其中,V2O5-Pr6O11-Al2O3催化剂的比表面积最大,孔道规整度较高,表面可还原钒物种最多,乙苯初始转化率最高;而V2O5-CeO2-Al2O3催化剂不仅具有较好的还原性能和较多的表面可还原钒物种,且氧缺陷含量最多,在CO2-ODEB反应中较好地维持了V5+与V4+之间的氧化还原循环稳定性,在表现出较高初始活性的同时,具有最佳长周期稳定性。虽然Pr6O11掺杂能够较好地抑制催化剂积炭,但影响催化剂稳定性的关键因素是CO2-ODEB反应中V5+与V4+之间的氧化还原循环,而不是积炭。

2.3 催化剂的CO2-ODEB反应性能

2.4 稀土氧化物的作用机制


3 结论
