氧化钴纳米球与低载银的相互作用及其对炭烟氧化的影响
2022-03-18何文宇何欣阳陈龙文付名利叶代启
王 婧,何文宇,何欣阳,陈龙文, 张 俊,付名利, 2, 3*,叶代启, 2, 3
(1 华南理工大学 环境与能源学院,广东 广州510006; 2 广东省大气环境与污染控制重点实验室,广东 广州 510006; 3 挥发性有机物污染治理技术与装备国家工程实验室,广东 广州 510006)
随着社会经济发展和产业结构升级,移动污染源排放逐渐成为影响空气质量的重要因素之一。机动车尾气排放产生的炭烟颗粒物(PM)是大气中细颗粒物(PM2.5)的重要来源,其在引起雾霾天气的同时严重危害人类健康。“十四五”期间,我国环保部门强调要加强PM2.5和臭氧(O3)的协同控制,在此背景下开展机动车排放炭烟颗粒控制技术研究具有重要现实意义[1-2]。
炭烟氧化需要充足的反应时间及高温、富氧的反应条件,其反应温度通常在500~600 ℃范围内,而机动车在中低负荷状态下运行时的排气温度一般在400 ℃以下。因此,制备低温、高性能的炭烟氧化催化剂是机动车尾气排放控制技术的关键。在诸多催化剂中,贵金属催化剂(Pt、Au、Pd等)因具有优异的氧化性能和较宽的适用范围而备受关注和研究[3-4]。Wu等[5]通过在La2O2CO3纳米棒表面制备Au@La2O3核壳,使炭烟催化氧化反应的T50值达到375 ℃。相比于其他贵金属,Ag的工业属性强、成本低廉且催化活性优异,受到越来越多的关注。Hernández等[6]采用不同的Ag负载量合成了银改性的锰酸盐(LaSrAgMn)和铁素体(LaSrAgFe)钙钛矿,在低氧浓度(O2体积分数1%)条件下,LaSr25AgMn的T50值为449 ℃,LaSr25AgFe的T50值为509 ℃,并发现Ag的状态和粒径决定了钙钛矿的催化性能。然而,Ag在炭烟氧化反应中的作用还存在争议,这是因为Ag负载材料的催化性能不仅取决于贵金属的性质与粒径,还取决于载体的氧化还原性能以及贵金属与载体之间的相互作用等。通过选择合适的载体氧化物以及调节载体与金属的相互作用,使载体活性位点与贵金属之间产生协同效应,对提升Ag基催化剂的催化活性至关重要。过渡金属氧化物中的钴氧化物和锰氧化物价态丰富,具有较好的氧化还原性能,在多相催化反应中表现出优异的氧化活性[7-11]。Yang等[12]通过选择性溶解法合成了α-Mn2O3,该化合物具有大量的介孔和氧吸附空位,更有利于CO的吸附和扩散,从而增加了CO与表面活性氧结合的可能性,在模拟颗粒捕集器(cGPF)条件下表现出很高的CO2选择性。此外,在许多针对Ag基催化剂的研究中,为了获得良好的催化性能,催化剂中贵金属的负载量通常较高(质量分数大于5%),这增加了催化剂的制备成本[13-15]。因此,在炭烟催化氧化反应中,探究Ag与不同过渡金属氧化物之间的相互作用对炭烟氧化性能的影响机制、开发高性能和低贵金属负载量的经济型低温催化剂具有重要现实意义。
本文重点研究金属-载体相互作用对Ag/Co3O4和Ag/Mn2O3催化剂上炭烟氧化反应的影响。通过XRD、H2-TPR、XPS、TG、EPR等方法对制备催化剂的结构和还原性能、活性氧物质和氧空位含量、表面化学成分和氧化状态进行表征,分析Ag分别与钴、锰氧化物之间的相互作用及其对炭烟氧化反应活性的影响。
1 材料与方法
1.1 催化剂的制备
球状钴氧化物的制备:在70 mL乙二醇中加入12.5 mmol的Co(NO3)2·6H2O (99.95%金属基,阿拉丁试剂有限公司),剧烈搅拌2 h,再加入35 mmol尿素,搅拌1 h。将生成的酒红色溶液转移至100 mL含特氟龙内衬的不锈钢高压釜中,150 ℃条件下反应12 h,自然冷却至室温后离心,收集粉红色混合物。用去离子水和无水乙醇多次洗涤粉红色混合物,除去未反应物,80 ℃条件下烘干过夜。烘干样品置于管式炉中600 ℃煅烧3 h,升温速率为2 ℃/min。
球状锰氧化物的制备:将上述钴氧化物制备方法中的Co(NO3)2·6H2O换成Mn(NO3)2·4H2O(99.95%金属基,阿拉丁试剂有限公司),其余操作步骤一致。
Ag负载催化剂的制备(湿浸渍法):将1 g钴氧化物或锰氧化物分散于去离子水中,分别滴加4 mL的0.059 mol/L硝酸银溶液,持续搅拌4 h使其充分负载,80 ℃烘箱中干燥过夜,600 ℃煅烧3 h,升温速率为2 ℃/min。
1.2 催化剂的表征
X射线衍射(XRD)测试在D8 ADVANCE X射线衍射仪(德国Bruker公司)上进行。该衍射仪采用Cu Kα辐射源,X-光管的工作电压为40 kV,工作电流为40 mA,扫描范围为10°~90°,扫描速度为8(°)/min。此外,还测试了扫描范围为30°~50°、速度为2(°)/min的慢扫。
N2吸脱附测试在ASAP 2460型物理吸附仪(美国Micromeritics 公司)上进行。先将样品于250 ℃真空条件下预处理6 h,再在-196 ℃条件下静态吸附N2。采用Brunauer-Emmett-Teller (BET)方法计算样品的比表面积,利用BJH方法分析样品的孔径分布。
使用JSM-7500F型场发射扫描电子显微镜(FESEM,日本日立JEOL公司)观察所制备催化剂的形貌和微观结构。
使用AutoChem Ⅱ 2920化学吸附仪(美国麦克默瑞提克仪器有限公司)进行氢气程序升温还原(H2-TPR)测试,表征所制备催化剂的氧化还原能力。称取50 mg样品置于U型石英管中,300 ℃氩气预处理1 h;待样品降至室温后,将气氛切换成体积分数为10%的H2/Ar混合气体(30 mL/min),吹扫至基线稳定;以10 ℃/min的恒定速率升温至800 ℃。通过TCD检测器检测H2消耗量,得到催化剂的H2-TPR曲线。
使用EscaLab 250Xi型X射线光电子能谱仪(美国赛默飞世尔科技公司)进行X射线光电子能谱(XPS)测试,分析所制备催化剂的表面元素价态。以Al Kα(hν=1 486.8 eV)为激发源,以C 1s峰(hν=284.6 eV)为参考,标定结合能值。
电子顺磁共振(EPR)在JEOL-fa300光谱仪上进行。测试温度为-196 ℃,频率为9.85 GHz,电源为10.8 mW。g值由频率和磁场强度决定。
耗氧再生测试在STA 449同步热分析仪(德国Netzsch 公司)上进行,步骤如下:400 ℃、N2气氛下加热30 min,升温速率为10 ℃/min;切换成体积分数为5%的H2,加热30 min;切换为空气气氛,保持30 min。
使用电感耦合等离子体发射光谱(ICP-OES,Perkin-Elmer Plasma 8000,USA)测定催化剂的化学成分。
1.3 催化反应性能评价
催化剂样品的催化活性评价主要通过炭烟程序升温氧化(soot-TPO)实验完成,具体步骤为:将制备催化剂与炭烟(Printex-U,Degussa,Germany)以10∶1的质量比混合2 min并研磨5 min,使催化剂与炭烟紧密接触,以该方式制得的样品可用于深入研究催化剂的内在活性[16]。为避免测试过程中的传质传热影响,将上述研磨混合物与石英砂以约1∶3的质量比混合,置于两团石英棉之间装入反应系统。通入体积分数为10%的O2/N2气体,气体流速为100 mL/min,空速(WHSV)为120 000 mL/(g·h),反应器温度以2 ℃/min的速率升至200~550 ℃。采用配备FID的在线气相色谱仪分析炭烟氧化产物CO和CO2的浓度,利用T10(着火温度)、T50(转化率为50%时的对应温度)和T90(转化率为90%时的对应温度)评价催化剂的催化活性。由于CO也是污染物,因此催化剂的CO2选择性也是本研究的考核指标。
2 结果和讨论
2.1 催化剂的结构与物理性质
采用X射线衍射(XRD)、氮气吸脱附和扫描电镜(FESEM)测试对所制备钴、锰氧化物催化剂的结构和物理性质进行表征。样品的XRD谱图如图1所示。在图1a中,Co3O4和Ag/Co3O4的衍射峰均表现出典型的钴基尖晶石相(PDF#73-1701)结构的特征反射,位于约18.9°、31.2°、36.8°、38.6°、44.8°、55.7°、59.3°、65.3°的衍射峰分别对应于Co3O4的 (111)、(220)、(311)、(222)、(400)、(422)、(511)、(440) 晶面[17]。以上结果表明,负载Ag后Co3O4未发生晶相转变,其衍射峰强度大,结晶度高。通过对Mn2O3样品进行分析,推断Mn2O3中的锰氧化物主要为立方晶体结构的Mn2O3(PDF#71-0636)。负载Ag后的Ag/Mn2O3样品出现了微量的四方相MnO2和AgOx的衍射峰,且结晶度变高, 推测Ag与Mn2O3之间存在一定反应。
为进一步考察Ag在2种氧化物上的状态,对Ag/Co3O4和Ag/Mn2O3进行30°~50°慢扫XRD表征,结果如图1b所示。图1b中位于38.1°和44.3°的衍射峰分别归属于金属Ag (PDF#87-0717)的(111)和(200)晶面,表明Ag纳米粒子(nanoparticles, NPs)在Co3O4上成功负载且高度分散。

图1 催化剂样品的XRD谱图Fig.1 XRD patterns of the catalyst samples
催化剂的N2吸脱附等温线和相对孔径分布结果如图2所示。所制备催化剂均表现出Ⅱ型吸附等温线(图2a),催化剂的回滞环符合H3型特征。结合孔径分布结果(图2b)可以看出,催化剂的内部孔隙以中、大孔为主(大于30 nm)。
由催化剂样品的FESEM结果(图3)可以看出,所有催化剂均呈现出规整的球状结构,均一性良好,负载Ag没有改变催化剂的形貌。对比发现,锰系催化剂表面更加粗糙,孔隙更多,能够明显看到催化剂表面的孔隙通道,这与氮气吸脱附表征结果(表1)相吻合。
对负载Ag催化剂Ag/Co3O4和Ag/Mn2O3进行Ag元素映射分析(EDS),结果可以清楚地看到Ag纳米粒子在钴、锰氧化物上分散性良好(图4)。
此外,还通过ICP-OES测定了催化剂样品的实际载银量,具体数据如表1所示。

V代表孔体积,D代表孔径。

2.2 催化剂的炭烟燃烧活性
图5和表2展示了催化剂样品的炭烟燃烧活性。在没有任何催化剂的情况下,空白炭烟燃烧反应在较高温度下开始,其T50值为515 ℃。引入过渡金属氧化物Co3O4和Mn2O3后,炭烟燃烧活性显著提升,Co3O4的T50值降至381 ℃, Mn2O3的T50值降至368 ℃。负载贵金属Ag后,2种过渡金属氧化物的炭烟燃烧性能产生巨大差异。Ag的加入可以显著促进Co3O4对炭烟的氧化,其T50值下降近100 ℃,但负载Ag对Mn2O的炭烟氧化性能几乎没有影响,说明Ag与Co3O4之间存在金属-载体强相互作用。

图4 负载Ag催化剂的Ag元素映射结果Fig.4 Ag elemental mapping results of the Ag loaded catalysts
对气固相反应而言,催化材料的比表面积通常对催化活性具有较大影响,但由表1中催化剂的理化参数可知,本研究中催化剂的比表面积与催化活性之间无正相关性。这是因为在炭烟燃烧过程中,反应的催化活性并不是由催化剂的表面积决定,而是取决于炭烟与催化剂的有效接触位点,这一现象与前人的研究结果相契合[18-20]。
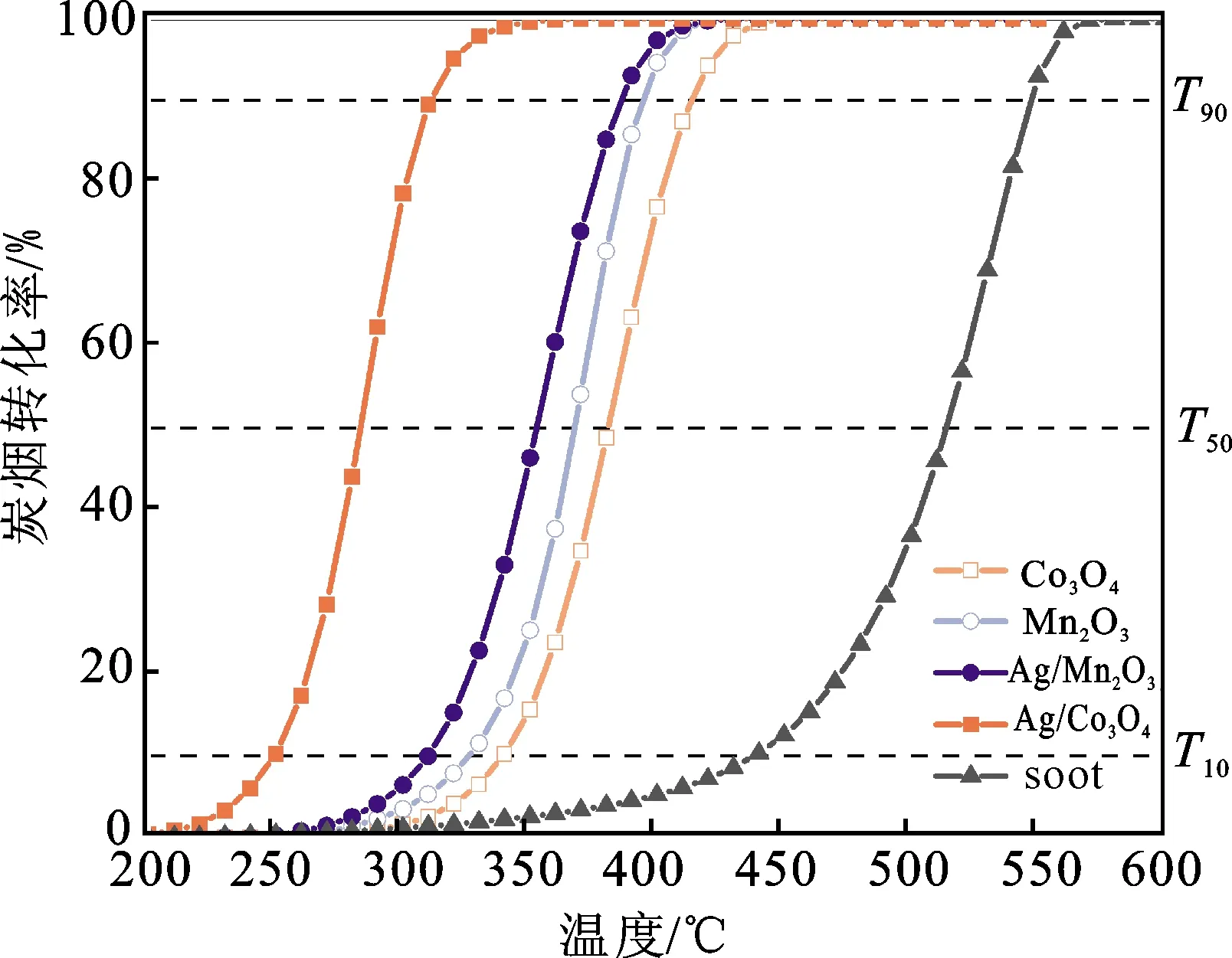
图5 催化剂样品的炭烟转化率Fig.5 Soot conversion of the catalyst samples
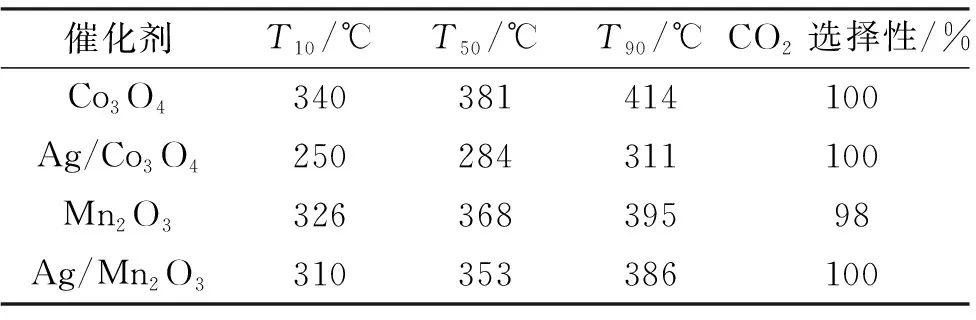
表2 催化剂样品的炭烟燃烧性能Tab.2 Soot combustion performance of the catalyst samples
2.3 Ag与钴氧化物之间的强相互作用
催化剂的还原性能对炭烟燃烧反应具有重要影响,因此采用H2-TPR测试对制备催化剂的还原性能进行表征,结果如图6所示。Co3O4样品在250~400 ℃范围内有1个宽峰,该峰实际包含有2种不同类型的还原峰,属于Co3O4中Co3+→Co2+→Co0的连续还原过程[21-22]。Mn2O3样品表现出2个还原峰,分别对应于Mn2O3还原为Mn3O4和MnO。当Ag 纳米粒子负载到钴、锰氧化物表面时,所有还原峰都向低温移动,如Ag/Co3O4比Co3O4的还原温度低195 ℃,这是由Ag与载体之间的相互作用引起的,表明Ag负载增强了样品的还原性。此外,Ag/Mn2O3位于200 ℃附近的低温肩峰可能是由AgO还原为金属Ag所致,金属Ag的溢出效应促进了Mn2O3还原,其H2活化能力高于裸露的氧化物表面[23];但引入Ag后Mn2O3的活化性能并没有显著提升,所以该低温肩峰也可能是Mn3+离子的非同步还原所致[12]。虽然Ag/Mn2O3在较低温度下开始还原,但Ag/Co3O4的低温耗氢量(13.8 mol/g)远大于Ag/Mn2O3(6.7 mol/g)。H2消耗量可以间接反映200~500 ℃范围内催化剂表面氧的消耗量,结合上述结果,说明催化剂的炭烟氧化活性强烈依赖于其表面的活性氧物种。
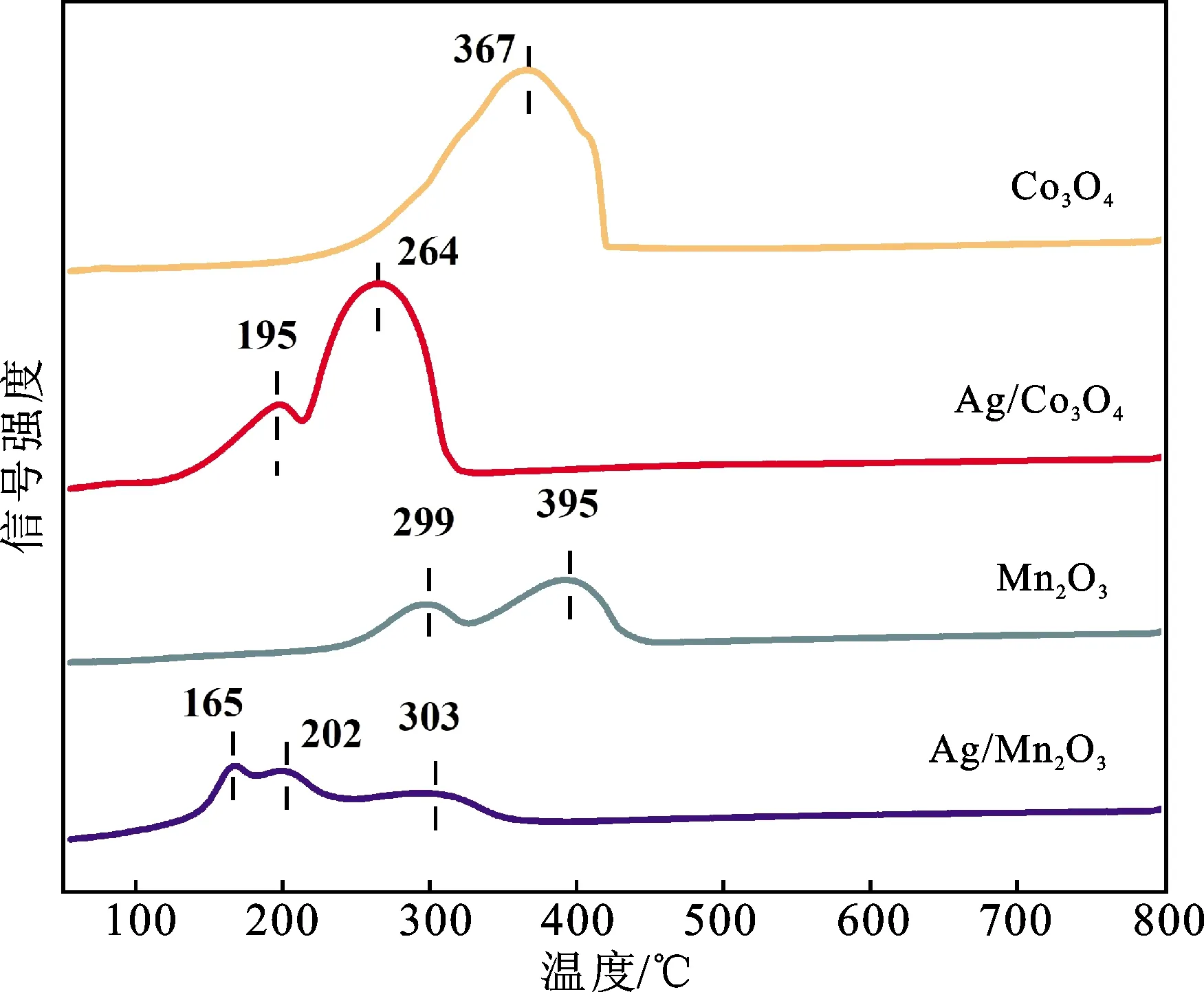
图6 催化剂样品的H2-TPR结果Fig.6 H2-TPR profiles of the catalyst samples
为进一步探究催化剂的氧化还原能力,在400 ℃等温条件下的热重反应中,通过来回切换还原和氧化气氛表征Ag/Co3O4和Ag/Mn2O3的氧消耗-再生能力[24]。由图7可以看出, Ag/Co3O4的质量损失在2个H2耗氧阶段都几乎是Ag/Mn2O3的2倍,说明Ag/Co3O4中氧的迁移率和消耗量更高。将反应气氛切换至体积分数为21%的O2时,Ag/Co3O4可以恢复到初始质量,而Ag/Mn2O3存在质量损失。这表明Ag/Co3O4具有更强的耗氧再生能力,说明Ag与Co3O4之间的强相互作用有利于晶格氧活化,能够促进Co3+/Co2+的氧化还原循环,从而使Ag/Co3O4表现出更高催化性能。这与H2-TPR测试中Ag/Co3O4还原性能最佳的结果一致。

图7 负载Ag催化剂的氧消耗-再生能力Fig.7 Oxygen consumption-regeneration of the Ag loaded catalysts
为明确催化剂中氧空位的含量,利用EPR响应对带有未配对电子的氧空位进行检测,结果如图8所示。锰氧化物在负载Ag后氧空位有所增加,对应的催化性能也略有提高,这是因为Ag可以促进活性氧物种Ox-的形成。与此同时,虽然锰氧化物的氧空位浓度明显高于钴氧化物,但其反应活性反而低于估氧化物。先前有报道指出,催化剂的氧空位对炭烟燃烧具有促进作用,但氧空位浓度过高会使活性氧物种在溢流到炭烟表面前就发生O2-→O-→O2-转变[19, 25],从而降低活性氧物种Ox-的利用率。因此,适量氧空位浓度更有助于炭烟氧化活性的提高,Ag/Co3O4表现出更高氧化性能。
最后,采用XPS技术探究Ag负载对Co3O4和Mn2O3表面化学状态的影响。如图9a中的O 1s XPS谱图所示,催化剂在532 eV附近出现有归属于羟基氧(Oγ)的峰,在530~531 eV和528~529 eV分别出现有归属于表面吸附氧(Oα)和晶格氧(Oβ)的峰[26-29]。根据H2-TPR表征的耗氢量结果(图6)及XPS测定的催化剂表面元素组成结果(表3)可知,Ag/Co3O4样品的活性氧含量最高,该结果与催化剂的炭烟氧化性能结果一致[28,30]。


图9 催化剂样品的XPS谱图Fig.9 The XPS spectra of the catalyst samples
在图9b中,Ag/Co3O4的Ag 3d5/2峰位于368.2 eV处,与Ag0物种有关。Ag 3d3/2和Ag 3d5/2在6.0 eV裂解下的XPS谱图进一步证实了Ag在催化剂上的金属性质。Ag/Mn2O3的Ag 3d5/2非对称峰预示着氧化态Ag的存在[31],该峰可细分为2个峰,其中,高结合能峰(367.7 eV附近)归属于Ag0,低结合能峰(368.5 eV附近)归属于Agδ+[32],这与前文XRD的表征结果一致。有研究认为,在炭烟催化氧化过程中,Ag0是催化活性组分,表面AgOx比例过高不利于炭烟起燃活性的提高[33]。此外,Ag/Co3O4催化剂具有更高结合能(368.2 eV),这可能归因于金属-载体之间的强相互作用;结合Co3O4负载Ag后Co 2p3/2峰的正向移动(图9c),推断电子从钴物种向Ag物种传输[14]。综上,Ag/Co3O4具有高活性的原因可能是金属-载体强相互作用加强了Ag与Co3O4界面的电子传递。
图9c为钴氧化物催化剂的Co 2p XPS谱图。位于Co 2p3/2的780.3 eV和Co 2p1/2的795.5 eV处的低结合能峰对应于Co3+物种,而位于Co 2p3/2的782.1 eV和Co 2p1/2的797.5 eV处的高结合能峰对应于Co2+物种。负载Ag后,Co3+/Co2+摩尔比增加,说明Ag能够促进钴物种从Co2+转变为Co3+。上述现象可以用Ag++ Co2+→Ag0+ Co3+来解释,在Ag/CeO2中也观察到了类似现象[14, 19, 34-35]。
图9d为锰氧化物催化剂的Mn 2p XPS谱图。Mn2O3和Ag/Mn2O3的Mn 2p峰具有相同结合能,表明二者的表面锰成分相同。位于641.3~642.1 eV和653.3~653.6 eV的2个峰分别对应于Mn 2p3/2和Mn 2p1/2。Mn 2p3/2的反褶积进一步说明催化剂表面存在3个Mn物种[36-39]。位于640.7~641.0 eV、641.9~642.1 eV和643.1~643.5 eV的峰分别对应于Mn2+、Mn3+和Mn4+。负载Ag后,Mn2O3和Ag/Mn2O3的Mn4+/Mn3+摩尔比不同,这可能是催化剂表面Mn3+部分转化为Mn4+所致。先前研究发现,伴随着氧化还原驱动的Mn2+和Mn4+氧化态的增加,可能会形成氧空位[40],这与本研究中负载Ag后Ag/Mn2O3的氧空位含量明显增加的结果(EPR表征)一致。
3 结论
采用水热法合成了球状钴、锰氧化物载体,并通过简单浸渍法将低含量贵金属Ag成功负载至钴、锰氧化物载体上。Co3O4负载Ag后,Ag/Co3O4的催化性能显著提升,表现出优于Ag/Mn2O3的最佳炭烟氧化活性。经H2-TPR、XRD、XPS等表征发现,Ag/Co3O4优异的炭烟催化燃烧性能归因于Ag与Co3O4之间的金属-载体强相互作用,这种相互作用增加了催化剂表面活性氧物种的数量,有利于氧物种的迁移,提升了催化剂的低温还原性能。本研究结果为低贵金属负载量的经济型低温催化剂的开发应用奠定了理论基础。
