稀土异相纳米催化剂的控制合成及应用研究进展
2022-03-18王德九张亚文
王德九,张亚文
(北京分子科学国家研究中心,稀土材料化学及应用国家重点实验室, 北京大学 化学与分子工程学院,北京 100871)
稀土(rare earth,RE)元素是元素周期表中15种镧系元素加上Sc、Y共17种金属元素的总称。在自然界中,稀土元素主要富集在花岗岩、碱性岩等矿物的矿床中[1-2]。通常而言,稀土元素以氧化物或含氧酸盐的形式与其他金属元素如Ca、Sr、Zr等共存于矿物中。稀土元素普遍具有较低的氧化还原电势。除了少数元素如Ce、Pr、Sm外,大部分稀土元素最稳定的氧化态为+3价。与其他常见的+3价金属元素如Al、Fe等相比,稀土元素的阳离子具有更大的离子半径和相对更弱的路易斯酸性,这使得稀土材料具有与Al、Fe等常见元素材料不同的结构和性能。另外,稀土元素具有丰富的4f电子能级,从而赋予了其独特的电子[3-4]、光学[5-8]和配位[9-11]性质,拓展了稀土材料的研究和应用空间。
稀土有工业“黄金”之称,以稀土元素为基础的材料拥有优良的光学、电学、磁学性能以及催化性能,是构建各种新型功能材料的基础,在现代工业中有着不可替代的作用[12-15]。在催化领域,稀土元素因具有特殊的4f电子能级以及较大的离子半径,展现出与众不同的催化性能,既可以在纳米异相催化剂中作为主要活性元素发挥作用(主要包含稀土氧化物和稀土合金),也可以定量地掺杂于其他催化材料中作为助催化剂来调控催化材料的性能。此外,随着近年来合成化学和催化化学的发展,稀土单原子催化剂也逐渐进入研究者的视野。相较于传统的贵金属催化剂,稀土催化材料成本低廉,应用前景广阔[16-17],因此得到研究者的广泛关注和研究。
不同类型的稀土纳米催化材料具有不同的合成策略。例如,稀土氧化物的合成常以稀土氯化物或稀土硝酸盐为前驱体,利用沉淀法或热分解法将其转化为氧化物形式,在此过程中可以很方便地将其他金属元素引入催化剂中,用来构造负载型催化剂。而稀土合金则可以借用合金工业中的成熟工艺,以稀土金属为原料,用熔铸法制备。与传统的异相催化剂相比,利用先进的纳米合成技术制备的纳米异相催化剂具有更大的比表面积,通常表现出更优异的催化活性。稀土异相催化剂也常常制备成纳米异相催化剂,以提高其性能并降低成本。合成化学的发展为稀土催化材料的合成提供了更广阔的思路,电化学方法、非水溶剂方法等新型稀土纳米催化剂合成手段的发展大大拓展了稀土异相纳米催化剂的应用空间。
文章从稀土异相纳米催化材料中稀土元素的存在形式和活性成分入手,将稀土异相纳米催化剂分为稀土氧化物、稀土合金、稀土助催化剂和稀土单原子催化剂4类(图1)。在此基础上,分别介绍了各类稀土异相纳米催化剂的结构特征和合成方法,并总结了稀土异相纳米催化剂在CO氧化、CO2加氢、水汽变换(WGSR)等C1转化反应和氢析出反应(HER)、氧还原反应(ORR)等电催化反应中的最新研究进展。最后,对稀土异相纳米催化剂的发展前景进行了分析和展望。

图1 稀土异相纳米催化剂的分类和应用Fig.1 Classification and application of heterogeneous rare earth nanocatalysts
1 稀土氧化物纳米催化材料
稀土元素普遍具有较低的氧化还原电势,催化环境下大多以氧化物或含氧酸盐的形式存在。因拥有比常见过渡金属更大的离子半径以及丰富的4f电子能级结构,与其他常用于催化反应的氧化物如Al2O3、Fe2O3、TiO2等相比,稀土氧化物催化剂具有更独特的配位性能以及光学、电学和磁学性能,拓展了其在电催化、光催化等领域的应用[18-19]。
按照稀土元素的价态,可以将稀土氧化物分为+3价氧化物和+4价氧化物。除CeO2、PrO2和TbO2外,大多数稳定的稀土氧化物都是+3价。不同氧化态的稀土氧化物具有不同的结构和催化性能。
1.1 稀土氧化物的结构
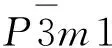


图2 稀土氧化物晶体结构示意图Fig.2 Schematic illustration of the crystal structures of rare earth oxides
RE2O3的相图如图3所示。其中,RE2O3(RE=La—Pr)具有A型结构;RE2O3(RE=Y,Dy—Lu)具有C型结构;其余RE2O3在低温下为C型结构,在高温下为B型结构。
目前为止,研究人员已经制备出许多具有明确形貌的稀土氧化物纳米材料,包括0D的纳米球、纳米立方体和纳米八面体,1D的纳米棒和纳米线,2D的纳米片和纳米带,3D的树枝状和花状结构等(图4)。不同形貌的稀土氧化物材料可能会暴露不同的晶面,进而对催化活性产生影响。Konsolakis等近期综述了CeO2纳米材料的形貌对各种非均相反应的影响[21]。借助各种化学合成方法调控稀土氧化物的形貌是调控稀土氧化物催化剂催化性能的有效策略之一。

图3 RE2O3的相图Fig.3 Phase diagram of RE2O3注:修改自文献[20]。

a. CeO2纳米空心球[22],b. CeO2纳米立方体[23] ,c. CeO2纳米八面体[24] ,d. CeO2纳米线[25] ,e. Yb2O3纳米棒[26] ,f. Eu2O3纳米管[27] ,g. Sm2O3纳米盘[28],h. La2O3纳米板[29] ,i. CeO2纳米板[30] ,j. Dy2O3花状球[28] ,k—l. CeO2纳米枝状晶[31-32]。
1.2 稀土氧化物纳米催化剂的合成
传统的稀土氧化物催化剂主要以稀土氯化物或含氧酸盐为原材料,通过简单的沉淀法或热分解法合成。传统合成方法往往使用低比表面积的前驱体在500 ℃以上的高温下煅烧得到催化剂产品,其得到的材料通常具有较小的比表面积和较低的表面缺陷密度,这对于催化剂而言是不利的。得益于合成化学的飞速发展,研究者已经开发出多种方法来合成高比表面积的稀土氧化物纳米催化剂,目前较为先进的稀土氧化物纳米材料合成方法包括溶剂热法、溶胶-凝胶法和金属有机框架(MOF)热解法等。此外,在实际应用中,常将稀土氧化物与其他金属或金属氧化物复合,制成负载型催化剂,因此将稀土氧化物纳米材料与其他金属复合的负载方法也非常重要。
1.2.1 溶剂热法
溶剂热法指在密闭体系如高压釜中,以水或有机物如乙醇、二甲基甲酰胺等作为溶剂,在溶液的自生压力作用下,令反应物在超过溶剂沸点的较高温度下进行反应的合成方法。溶剂热法兼具溶液中前驱体均匀混合的优点和固相反应中高反应温度的特点,有利于合成生长缺陷少、取向好的晶体;其合成的产物结晶度高且易于控制产物晶体的形貌和粒度,非常适合用于模型催化剂的制备。
本课题组前期利用水热法合成了CeO2纳米线。该研究在前驱体中加入适量NaCl,借助Cl-与Ce(OH)3晶面的选择性配位,实现晶体各向异性的生长,从而得到尺寸均一、结晶良好且主要暴露催化活性较高的(110)晶面的CeO2纳米线(图5)[25]。

图5 掺入Nd、Lu的CeO2纳米线 HAADF-STEM EDS元素分布图像Fig.5 HADDF-STEM EDS elemental mapping of CeO2∶Nd and CeO2∶Lu nanowires注:参考自文献[25]。


图6 阴离子配位下氧化铈纳米棒 到纳米立方体的转化Fig.6 Conversion of cerium oxide nanorods to nanocubes with anion coordination注:修改自文献[34]。
1.2.2 溶胶-凝胶法
溶胶-凝胶法是以金属盐或金属酯类化合物为前驱体,与其他配体混合均匀后,在一定温度下反应形成凝胶,最后经干燥、灼烧等后处理得到氧化物的合成方法。在稀土氧化物的制备过程中,金属硝酸盐常被用作前驱体,聚乙烯吡咯烷酮(PVP)、柠檬酸盐等常被用作配体。溶胶-凝胶法的反应条件相对温和,很容易实现多元素的均匀掺杂,得到的产物往往具有较大的比表面积,是一种合成多元稀土氧化物的有效方法。
Xiao等利用溶胶-凝胶法,以柠檬酸作为配位剂,将第三种金属M(M=La,Tb,Zr)掺杂到Ni-CeO2催化剂中[36]。不同元素掺杂得到的纳米颗粒均表现出Ce和M的高度均匀性,但Ni的分散性具有差别,进而在催化性能上产生差异。Chávez-garcía等利用溶胶-凝胶法制备了Eu、Bi掺杂的Y2O3纳米颗粒,掺杂元素同样具有均匀的元素分布[37]。
溶胶-凝胶法还可以用于高度复杂体系的合成。例如,Kim等利用EDTA-柠檬酸盐作为络合剂,制备得到Ce1-x(TM,RE)xO2-δ催化剂;该合成方法有效避免了掺杂组分的相分离,并可以获得粒径较小的纳米颗粒[38]。Riley等利用溶胶-凝胶法,将4种及以上金属元素以较高的比例掺入CeO2晶体,成功制成高熵氧化物(high entropy oxide, HEO);该方法在CeO2晶格中引入高比例过渡金属的同时保持了CeO2晶体结构的稳定性[39]。与直接将前驱体混合并煅烧的方法相比,溶胶-凝胶法制得的催化剂具有更小的尺寸和更粗糙的表面,从而表现出更优的催化活性。
1.2.3 金属有机框架(MOF)热解法
碳基材料和杂原子的引入可以保护金属活性位点并增强其电荷传输性能,是设计与合成非贵金属基电催化剂的有效策略。MOF可以提供碳骨架和杂原子,同时还具有可调控的组成、巨大的孔隙率和较高的比表面积。因此,MOF热解法既可以用来合成含有碳及杂原子的稀土催化剂,也可应用于向其他金属催化剂中引入稀土组分,是一种行之有效的合成方法。通过合理且精细的热解条件控制,可以维持MOF材料的高比表面积并得到孔道结构的氧化物材料,这对提高催化剂的原子利用率具有重要意义。
在Xiong等的研究中,利用ZIF-8作为结构模板,通过离子交换法制备出负载在碳布上的CeO2-CoP-C纳米催化剂,并将其用于电催化氢析出(HER)反应(图7)[40]。与未掺入CeO2的样品相比,CeO2-CoP-C表现出更高的反应活性和更低的过电势。 进一步的结构表征表明该催化剂具有和

图7 CeO2-CoP-C/CC形成示意图Fig.7 Schematic diagram of CeO2-CoP-C/CC formation注:修改自文献[40]。
ZIF-8前体相当的孔道尺寸和高于传统CeO2催化剂的比表面积,能够提供更丰富的反应位点并有利于提高催化剂中活性物质的转移速率,从而表现出优异的催化性能。
Zhang等以Ce-MOF为前驱体,成功制备出新型CeO2/Au@SiO2核壳纳米管催化剂(图8)[41]。其先通过简单的水热反应合成Ce-MOF,然后负载Au纳米颗粒,再用SiO2进行封装,最后将材料热解得到多孔CeO2/Au@SiO2催化剂。夹在CeO2载体和SiO2壳之间的Au纳米颗粒显示出与CeO2载体较强的相互作用,在4-硝基苯酚还原反应中表现出优异的催化性能。

图8 CeO2/Au@SiO2形成示意图Fig.8 Schematic diagram of CeO2/Au@SiO2 formation注:修改自文献[41]。
除了以上3种稀土氧化物纳米催化剂合成方法外,微乳法、模板法等其他方法也常常被用于稀土氧化物纳米催化剂的合成,目前常用的相关合成方法及其原料、反应条件、优势和缺陷总结于表1。
1.2.4 稀土氧化物负载型催化剂的合成
目前,已经有多种有效方法可以用来制备金属/氧化物负载型催化剂,其中最常用的方法有浸渍法、共沉淀法等,这些通用方法也适用于稀土氧化物负载型催化剂的合成。
浸渍法是制备负载型催化剂的常见方法。在利用浸渍法制备稀土氧化物负载型催化剂的过程中,将稀土氧化物载体与含有金属前驱体的溶液混合,通过调控溶液的化学环境,如金属前驱体的浓度、pH值、配位离子(如Cl-)的浓度等,令金属前驱体可控地通过物理或化学作用吸附在载体表面。浸渍法过程简单,反应条件易于控制,还可以在很大程度上保持载体形貌,适合模型催化剂的合成。

表1 稀土氧化物的合成方法Tab.1 Synthesis methods of rare earth oxides
本课题组前期利用浸渍法,以贵金属氯化物为前驱体,在CeO2纳米线上成功负载了Ru、Rh、Pd、Ir、Pt 5种贵金属,并通过调控前驱体的浓度和浸渍时间,成功实现了对不同贵金属尺寸的精确调控[42-43]。以Ru/CeO2为例,研究制备了3种不同尺寸的Ru/CeO2样品,分别为单原子Ru(SA)/CeO2、纳米团簇Ru(NC)/CeO2和纳米颗粒Ru(NP)/CeO2(图9)。HAADF-STEM(高角环形暗场扫描投射电子显微镜)图像表明:在Ru(NC)/CeO2和Ru(NP)/CeO2样品中,Ru颗粒的尺寸分别为(1.18±0.25)nm和(4.01±0.80)nm;而在Ru(SA)/CeO2样品中,Ru呈单原子分散。扩展X射线吸收精细结构谱表明Ru(SA)/CeO2样品不存在Ru-Ru配位,进一步佐证了Ru呈单原子分散的结构特点。
共沉淀法也是一种合成负载型稀土氧化物纳米材料的简便手段。共沉淀法通常需要将待负载的金属前驱体与稀土前驱体配制成均匀溶液,再加入合适的沉淀剂;通过在溶液中加入表面活性剂以及调整反应温度等方式,可以在一定程度上调控共沉淀产物的组成和形貌。Pudukudy等利用共沉淀法制备得到了Fe/La2O3和Fe/CeO2纳米材料[44]。其使用Fe和La、Ce的硝酸盐作为金属前驱体,碳酸铵作为沉淀剂,沉淀后在700 ℃条件下煅烧得到催化剂产物;该催化剂呈薄片状,具有疏松多孔的表面。Li等使用硝酸盐作为金属前驱体,草酸铵作为沉淀剂,合成了钙钛矿结构的BaCeO3,并将其当作Ru的载体用于合成氨反应[45]。通过在金属前驱体中加入La(NO3)3,可以很简便地在载体中掺入La,从而增加载体中的氧空位,进而提高催化剂的催化活性。
总体来说,经过多年的发展和积累,稀土氧化物的合成方法已经相当丰富,能够满足大部分催化剂的合成需求,但其仍有较大发展空间。例如,目前虽然可以通过浸渍法得到颗粒尺寸可控的贵金属负载型催化剂,但这种方法所制备催化剂的贵金属负载量比较低,导致稀土原子利用率低,不利于成本控制。MOF热解法虽然已经有相当多的研究和报道,但MOF的合成和热解条件比较复杂且缺少规律,保留MOF的孔道结构仍然比较困难;此外,MOF的合成成本高,也限制了MOF热解法在工业催化剂生产领域的应用。若上述问题能够得到有效解决,稀土氧化物催化剂的应用前景将会得到进一步拓展。
1.3 稀土氧化物的催化应用
以CeO2、La2O3为主的稀土氧化物具有优秀的热稳定性和O离子迁移性能,CeO2更是具有可逆储放氧的能力,因而稀土氧化物催化剂在各类热催化反应中都得到了广泛应用,例如,CO氧化反应、水汽变换反应、CO2加氢反应、甲烷重整反应等。尽管稀土氧化物普遍具有较差的导电性,限制了其在电催化、光催化领域的应用,但通过与其他材料复合,稀土氧化物同样可以在电催化和光催化领域发挥优势。稀土氧化物纳米催化剂在催化反应中的最新研究和应用进展主要体现在以下几个方面。

a、c. 球差校正的HAADF-STEM图像;b. Ru(SA)/ CeO2对应元素分布图;d. Ru(NC)/ CeO2对应元素分布图;e. Ru(NP)/ CeO2的HAADF-STEM图像;f. Ru(NP)/CeO2的HRTEM图像,其中的插图是Ru纳米颗粒的HRTEM放大图像,显示了六方Ru(101)晶格条纹。
1.3.1 CO氧化反应
CO氧化反应在汽车尾气处理、CO传感器、室内空气净化等领域应用广泛。此外,由于其简单的反应式和反应机理,也常被用作探针反应,用于研究催化剂的构效关系[46]。Langmuir-Hinshelwood机理和Mars-van Krevelen机理是目前最常见的2种CO催化氧化反应机理[47-50]。在Langmuir-Hinshelwood机理中,O2在催化剂表面吸附并解离成吸附态的O原子,再通过与吸附态的CO反应生成CO2产物。而在Mars-van Krevelen机理中,CO分子与金属氧化物的晶格氧发生反应,生成产物CO2分子并在原位产生一个氧空位,O2分子继而在氧空位附近吸附并解离,生成O原子并填补空位。在稀土氧化物催化剂的反应中,Mars-van Krevelen机理更为常见。根据Mars-van Krevelen机理的反应过程可知,催化剂表面的氧空位对催化反应至关重要,因此具有可逆生成氧空位能力的催化材料往往具有优秀的CO氧化反应催化性能。稀土氧化物材料中的O原子可以在晶格中快速迁移,有利于氧空位生成;同时,以CeO2为主的+4价稀土氧化物具有优异的变价性能,其晶格中本身就存在大量氧空位,且氧空位的含量可以随外界环境变化而迅速变化。因此,以CeO2为主的稀土氧化物是优良的CO氧化反应催化剂。
稀土氧化物材料可以独立地催化CO氧化反应。本课题组前期围绕CeO2基材料在CO氧化反应中的应用进行了系统且深入的研究,揭示了稀土元素掺杂对CeO2催化CO氧化反应的影响[25]。研究发现:掺杂样品的催化性能与掺杂元素的离子半径具有类“火山型”关系,其中掺杂Nd的样品表现出最好的反应性能;稀土元素掺杂会改变氧化铈表面的氧释放能力,轻稀土掺杂样品的氧空位形成能远低于重稀土掺杂样品;稀土掺杂还会影响表面吸附物种(即碳酸盐和碳酸氢盐)的键合构型,红外实验证明掺杂Nd的样品上存在最多的单齿碳酸盐中间物种,而这些中间物种可以迅速地转化为CO2产物。
CeO2的形貌调控对CO氧化反应也有着重要影响。Mofarah等开发了一种快速、灵活的无模板方法来合成介孔CeO2-x纳米材料,该合成材料具有高密度的缺陷和高达251 m2/g的比表面积,表现出极为优异的CO氧化性能(T90%=148 ℃)[51]。Lim等成功合成了一系列具有不同壳厚度的CeO2空心球,样品的CO氧化反应活性随着壳厚度的减小不断提高,作者认为壳层厚度减小引起的介孔率提高是导致CO氧化反应活性增强的主要原因[22]。此外,不同形貌CeO2暴露的晶面有所不同,而不同晶面具有不同的储氧行为和吸附性能,进而会产生不同的CO氧化性能[34]。Lustemberg等利用先进的DFT模拟方法和单晶红外光谱实验研究了在CeO2的(110)和(111)晶面上CO分子的吸附行为,发现在这2个晶面上,CO分子的吸附构型和吸附能都存在显著差别,这显然会影响CO氧化性能[52]。因此,合理的催化剂结构设计对高效催化剂的开发具有重要意义。
单一组分的稀土氧化物纳米催化剂对CO氧化反应的催化活性较为有限,并不能满足实际工业生产的需求。因此,常将稀土氧化物纳米催化剂与其他过渡金属复合制成负载型催化剂,这类催化剂同时具备稀土氧化物储放氧的能力和金属活性位点活化CO分子的能力,从而展现出非常优异的催化性能。近年来,本课题组对贵金属/CeO2负载型催化剂进行了深入研究,报道了局部配位结构对低温CO氧化中Pt/CeO2材料催化活性的影响;在密度泛函理论计算和原位光谱实验的辅助下,发现Pt纳米团簇中较低的Pt-O配位数有利于提高反应活性[53]。此外,课题组还对CeO2负载的不同贵金属催化剂在CO氧化中的粒径效应进行了研究,发现CO氧化反应在M/CeO2(M=Ru,Rh,Ir)上遵循Mars-van Krevelen机理,但在Pd/CeO2上遵循与Mars-van Krevelen机理不同的碳酸盐中间体反应机理,这也导致Pd与Ru、Rh、Ir表现出不同的尺寸效应(图10)[54]。诸多原位及非原位表征结果表明:催化反应路径和活性位点上的差异归因于M/CeO2(M=Ru,Rh,Ir)催化剂不同的金属载体电子相互作用,其中在CeO2负载的Rh亚纳米团簇催化剂上,Rh与CeO2之间的金属载体电子相互作用最有利于吸附的CO分子活化,因而表现出最佳的催化性能[54]。除贵金属催化剂以外的其他过渡金属也是有效的CO氧化反应催化剂,而且其成本远低于贵金属。例如,Kim等研究了同时掺杂稀土和过渡金属的CeO2催化剂在CO氧化反应中的催化活性和稳定性[38]。研究发现:稀土元素掺杂可以抑制催化剂烧结,提高催化剂的稳定性,而过渡金属掺杂可以通过促进表面缺陷的形成提高催化活性;其中,La、Sm、Cu共掺杂的CeO2具有最为优异的催化活性和良好的稳定性,其高活性可以在反应条件下维持700 h。
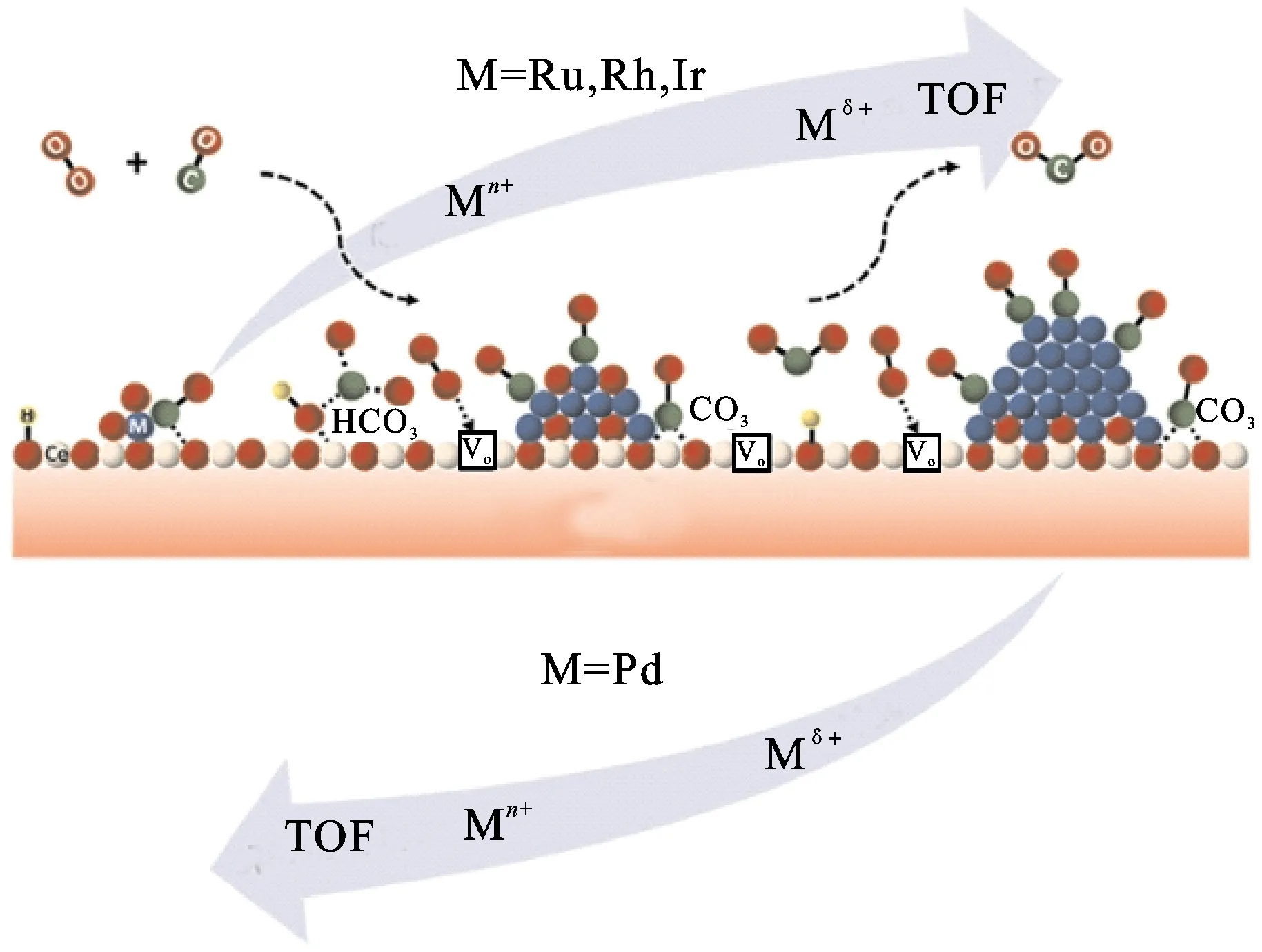
图10 不同贵金属/CeO2催化CO 氧化反应的构效关系示意图Fig.10 Schematic diagram of the structure-activity relationship of CO oxidation catalyzed by different precious metals on cerium oxide注:参考自文献[54]。
以稀土氧化物,尤其是CeO2为载体的负载型催化剂的催化性能同样受到载体形貌的影响。Lykaki等探究了负载在不同形貌CeO2上的Cu催化剂在CO氧化反应中的催化性能[20]。研究发现:负载在CeO2纳米棒上的Cu催化性能最佳。多种表征手段表明Cu/CeO2催化剂催化CO氧化反应的性能与Cu+及氧空位的含量密切相关,CeO2纳米棒上暴露的(110)晶面更容易生成氧空位,从而更有利于CO氧化。Li等比较了负载在不同形貌CeO2上的Ru/CeO2催化剂在低温CO氧化反应中的催化性能,发现负载在CeO2纳米棒上的Ru催化剂催化性能最优[55]。通常认为CeO2纳米棒会暴露更多的(110)晶面,但该研究借助先进的HRTEM表征阐明了CeO2纳米棒表面也会暴露具有丰富缺陷的(111)晶面(包含台阶、氧空位等),这些富含缺陷的(111)晶面与Ru形成强烈的金属载体相互作用,成为该催化剂性能优异的关键因素。
除了通常环境下的CO氧化反应外,在大量H2存在的环境中CO优先氧化(PrOx-CO)也有着重要应用,如除去合成氨原料气中的微量CO等。该反应对催化剂提出了新要求,需要催化剂在尽可能完全氧化CO的情况下,避免H2燃烧反应的发生。近年来,Au基催化剂在CO优先氧化反应中的优异性能逐渐得到研究人员的关注。相比传统的Pt基催化剂,Au基催化剂在保持较强CO活化能力的基础上,具有较弱的H2活化能力,表现出更高的CO2选择性,可以最大程度地保留原料气中的H2,更符合生产需求。例如,Sousa等开发了负载在SBA-15上的Au/CeO2催化剂[56]。与不加CeO2的Au/SBA-15相比,Au/CeO2/SBA-15催化剂将最大CO转化率的温度从300 ℃降低至100~135 ℃,CeO2与Au的金属载体相互作用在反应中起到至关重要的作用。Papavasiliou等制备了负载在CuCeOx载体上的Au单原子,并研究了后处理方法对其催化性能的影响[57]。研究发现该催化剂上存在高度分散的Au/CuOx物种,预还原过程会造成Cu物种表面富集,促进其与Au相互作用,从而表现出更强的CO优先氧化活性。除贵金属外,Cu基催化剂也常用于CO优先氧化反应。Marie等利用共沉淀法制备了Cu/Y2O3-ZrO2催化剂,并将其用于CO优先氧化反应,发现在1% CO、10.5% H2、1.5% O2的气体组成和250 ℃的反应温度下,该催化剂可以实现90%的CO转化率[58]。
总体来说,稀土氧化物基纳米催化剂中丰富的氧空位在CO氧化反应中扮演着重要角色,调节氧空位的策略包括掺杂、形貌调控等手段。除此之外,催化剂的比表面积、孔隙比例等因素也对其催化性能有着显著影响。由于稀土氧化物对CO分子的吸附和活化性能较弱,将稀土氧化物与过渡金属复合制成的负载型催化剂具有更广阔的应用空间,而对负载型催化剂的性能优化还需考虑负载金属的形貌、氧化态以及金属载体的相互作用等因素。
1.3.2 水汽变换反应
水汽变换反应(WGSR)在氢燃料电池和氨合成工业中至关重要,是生产氢气以及除去对许多工业催化剂有明显毒化作用的CO的有效手段。大多数水汽变换反应的催化剂为负载型催化剂,单一的稀土氧化物材料几乎不具有该反应的催化活性。但水汽变换反应需要催化剂同时具备活化CO分子和H2O分子的能力,且通常需要较高的反应温度,而稀土氧化物纳米材料由于具有良好的热稳定性和丰富的氧空位,在水汽变换反应中被广泛应用。
在各种催化体系中,贵金属/CeO2催化剂因其高稳定性和高活性被广泛应用于水汽变换反应。在诸多贵金属/CeO2催化剂中,Pt/CeO2催化剂是公认的在水汽变换反应中催化活性优异的催化剂。本课题组前期研究了Pt/CeO2催化剂在水汽变换反应中的尺寸效应[59]。该研究首先利用浸渍法合成了一系列高度均匀的Pt/CeO2催化剂,其中Pt颗粒尺寸覆盖了单原子、亚纳米团簇、纳米颗粒3个尺度;继而通过多种表征手段,揭示了Pt/CeO2尺寸效应背后的界面电子和几何效应对水汽变换反应的影响。Li等利用多种新型表征手段(原位X射线光电子能谱、原位透射电子显微镜等)对Pt/CeO2催化剂在水汽变换反应中的活性位点进行了细致研究,阐明了Pt/CeO2催化剂上水汽变换反应的活性位点和反应机理[60]。在上述反应条件下,Pt纳米簇中存在多种Pt氧化态,水汽变换反应最有可能通过界面处Pt0-氧空位-Ce3+位点的氧化还原路径完成(图11)。
Au/CeO2催化剂也是一种常见的水汽变换反应催化剂。Xiang等开发了中空介孔CeO2微球,并负载Au单原子,用于催化水汽变换反应[61]。该团队利用喷雾热解法制备了CeO2微球,该微球具有较大的介孔结构和高比表面积,非常有利于Au单原子负载,利用简单的浸渍法即可在其上负载Au单原子。相比于当前利用其他方法合成的Au/CeO2催化剂,Xiang等制备的催化剂表现出更好的催化活性和稳定性。Schilling等利用原位拉曼光谱、同位素实验以及DFT+U计算,阐明了Au/CeO2催化剂表面氧物种在水汽变换反应中的功能,揭示了Au/CeO2催化剂在水汽变换反应中的反应机理和氧原子迁移路径[62]。该研究观察到:在水汽变换的反应条件下,Au/CeO2催化剂中的CeO2载体被强烈还原,生成大量氧空位,表面的氧原子和羟基物种均可以与气氛中的H2O发生交换;此外,Au/CeO2催化剂表面的氧原子还可以迁移到晶格内。

红球和蓝球分别代表金属态和氧化态Pt原子,2个棕色球代表界面处的Pt活性位点。
除CeO2外,其他稀土氧化物在水汽变换反应中也有应用。Shi等利用两步法制备了Au/Pr(OH)x催化剂,并将其用于水汽变换反应和CO氧化反应[63]。该催化剂在2个反应中均显示出高活性、高稳定性以及比Au/CeO2更好的水汽变换反应活性。研究发现,Pr(OH)x载体具有大量的表面氧空位,有助于H2O解离,进而提高了水汽变换反应活性。
对于过渡金属/CeO2催化剂在水汽变换反应中的应用,Cu基催化剂报道较多,其他过渡金属也有报道。例如,Hu等近期利用简单的溶胶-凝胶法制备了一系列Cu/CeO2-ZrO2催化剂,并通过多种技术对其进行表征,发现氧空位浓度和Cu分散度是影响Cu/CeO2-ZrO2催化活性的重要因素[64]。上述材料在350 ℃下具有接近化学平衡极限的转化率和100%的CO2、H2选择性。Cámara等开发出基于Cu-CeO2的反向催化剂,即CeO2/CuO催化剂,并研究了向CuO中掺杂Mn、Zn对催化性能的影响,发现Mn、Zn掺杂会影响CuO上CeO2颗粒的粒径,进而对其催化水汽变换反应的活性产生重要影响[65]。Kim等研究了La、Pr、Zr掺杂对Co/CeO2催化剂催化水汽变换反应性能的影响;与Cu基催化剂类似,该研究中也观察到氧空位浓度和Co分散度对反应的催化性能具有重要作用[66]。这一现象对过渡金属/CeO2催化剂的深入研究和进一步发展具有指导意义。
稀土氧化物材料对CO的吸附较弱,活化O—H键的能力也较弱,因此无法单独催化水汽变换反应,而是常用作催化剂载体。稀土氧化物基纳米催化剂上丰富的氧空位以及较强的氧原子迁移能力对水汽变换反应具有关键作用,其氧空位对活性金属如Pt、Au的分散以及金属载体相互作用的调控也至关重要。
1.3.3 CO2加氢反应
CO2加氢反应根据反应条件的不同可能产生多种不同的产物,本节所讨论的CO2加氢反应泛指以CO2和H2为反应物,在一定温度下将CO2加氢生成还原产物的反应。CO2是主要的温室气体,将发电厂、炼钢厂等地产生的大量CO2转化为高附加值产品对缓解温室效应有着重要意义。然而,CO2分子的化学稳定性高且反应机理复杂,加氢反应可能产生多种不同反应产物,这对该反应催化剂的设计是一个巨大挑战。在CO2加氢反应中,稀土氧化物催化剂已经得到广泛研究和应用。与水汽变换反应类似,由于稀土氧化物本身难以活化CO2分子,对H2的吸附和活化也比较困难,单独的稀土氧化物并不能很好地催化CO2加氢反应。在应用以稀土氧化物为载体的负载型催化剂时,CO2加氢反应最常见的产物为CO和CH4,这2个反应分别称为逆向水汽变换反应和CO2甲烷化反应。根据CO2吸附构型的不同,上述反应可能通过碳酸盐中间体、甲酸盐中间体和CO中间体进行。当这些中间体的吸附能改变时,催化剂催化CO2加氢反应的选择性也会随之改变。通过掺杂、形貌调控等手段改变载体的电子结构和氧空位浓度,进而调节金属载体的相互作用,是调控CO2加氢反应性能的重要策略。
本课题组近期深入研究了贵金属/CeO2负载型催化剂在催化CO2加氢反应过程中的尺寸效应[42]。结果表明:在高活性Ru/CeO2催化剂上,金属载体强相互作用(SMSI)和H原子溢流效应对CO2甲烷化非常重要;改变CeO2载体上的Ru尺寸,SMSI和H原子溢流效应会随之改变,并对反应活性产生反向作用,导致反应活性与Ru尺寸呈现出“类火山型”关系;特定尺寸的Ru纳米团簇对CO2甲烷化具有最高催化活性(图12)。该研究阐明了Ru/CeO2催化剂在CO2甲烷化反应中的尺寸效应,并揭露了其机理,对高活性催化剂的设计具有指导意义。

图12 竞争性强金属载体相互作用和H溢流效应 对CeO2负载的Ru单原子、Ru纳米团簇和 Ru纳米颗粒上CO活化和表面脱水的影响示意图Fig.12 Competitive SMSI and H-spillover effects lead to CO activation and surface dehydration for CeO2-supported single Ru atoms, Ru nanoclusters and large Ru nanoparticles注:修改自文献[42]。
在此基础之上,本课题组又研究了CeO2负载不同贵金属催化剂的金属载体相互作用对CO2加氢反应活性和选择性的影响[43]。研究发现由于相互作用的金属载体不同,反应机理也有所不同,进而导致反应选择性(CH4/CO)存在差别;说明通过改变负载型催化剂中活性金属的成分,可以显著调节有效金属载体的相互作用,进而调节催化反应性能和产物选择性。
Cao等利用CeO2缺陷实现了对Pd/CeO2负载型催化剂的尺寸控制,并在此基础上研究了Pd尺寸对逆向水汽变换反应的影响,发现较大尺寸的Pd颗粒具有较高的电子密度,导致其具有更强的H2解离能力和H溢流效应,进而表现出较高的反应活性[67]。Xu等研究了Cr掺杂在Ru/CeO2催化剂催化低温CO2甲烷化反应中的影响[68]。该研究表明:在Ru/CeO2催化剂上,低温条件下CO2甲烷化以甲酸盐路径为主。CO2首先与表面羟基相互作用产生碳酸氢盐物种,碳酸氢盐继而被转化为甲酸盐,并进一步被氢化为CH4。Cr的引入增加了表面氧空位和羟基含量,促进了碳酸氢盐及甲酸盐的生成,因此Cr掺杂极大提高了Ru/Ce0.9Cr0.1Ox催化剂在低温下的活性。
非贵金属催化剂中,Ni基催化剂在CO2甲烷化反应中的应用最为常见。Yang等利用多种手段深入研究了稀土掺杂的Ni基介孔Ce0.8Zr0.2O2固溶体催化剂,发现稀土元素掺入可以显著提高催化剂的表面碱度,增强催化剂对CO2的化学吸附;此外,稀土元素还可以调控载体的电子结构,促进CO2分子活化[69]。Zhu等探究了Y掺杂的Ni/CeO2催化剂在CO2甲烷化反应中的性能,发现Y的引入可以促进氧空位产生,有利于CO2分子解离,从而使掺杂后的样品表现出比Ni/CeO2更好的CO2甲烷化活性[70]。
传统CO2加氢反应的主要产物通常为CH4或CO,但随着该领域的发展,通过CO2加氢得到甲醇的反应得到越来越多的关注。目前实现该反应主要是通过合理的催化剂设计抑制CO2过度加氢生成甲烷,并通过较低的反应温度和较高的反应压力抑制逆向水汽变换反应。工业中通常使用Cu/ZnO-Al2O3催化剂、在5~10 MPa高压下实现上述反应[71],针对该反应提高催化性能的策略之一就是改进Al2O3载体。近期,Zhu等报道了通过单步火焰喷雾热解方法制备的Cu/ZnO-CeO2催化剂,并研究了其CO2加氢性能[72]。该催化剂在3 MPa压力下具有与市售催化剂相当的反应活性,同时对甲醇的选择性提高了约60%。其研究团队指出,Cu-CeO2催化剂凭借较高的甲酸盐中间体覆盖度和更高的CO中间体加氢速率抑制了CO分子的直接脱附,进而提高了对甲醇的选择性。另外,Rezvani等系统地研究了Au/CeO2催化剂上CO2加氢合成甲醇的反应机理[73]。该研究通过动力学测量、稳态同位素瞬态动力学分析(SSITKA)、红外光谱(DRIFTS)等表征手段发现:在反应的初始阶段,催化剂表面会逐渐累积碳酸盐物种并阻塞活性位点,随着活性位点被阻塞,相比于甲醇的生成,逆向水汽变换反应会被更明显地抑制,导致甲醇选择性随着运行时间的延长而增加;此外,表面甲酸盐物种会在后续反应中转化为甲醇,说明甲酸盐物种是CO2加氢生成甲醇的重要中间体。
在已有研究的基础上,研究者还进一步利用掺杂等手段优化了CeO2载体。例如,Sharma等研究了In作为助催化剂对Cu/CeO2催化剂在CO2加氢制甲醇反应中的影响,发现In的引入显著增加了铜的分散性并降低了铜粒径,从而提高了催化活性;In的引入还增强了Cu和CeO2之间的金属载体相互作用,使铜颗粒在反应过程中更加稳定[74]。
除了传统的热催化领域外,CeO2基催化剂还可以被应用于CO2的光热还原。Ullah等报道了Co/CeO2催化剂在CO2甲烷化反应中的光效应[75]。研究表明:在250~350 ℃的温度范围内,光照能够有效提高上述反应的催化活性;其中,蓝光(450~460 nm)照射可以使催化剂活性提高116%,并表现出94%的CH4选择性。该团队将观察到的上述优异性能归结为CeO2基催化剂中的固有氧空位[75]。
CO2加氢反应的反应产物和反应机理都相当复杂,在催化剂的选择和设计上必须考虑副反应对目标产物产率的影响。在CO2加氢反应中,酸性分子CO2的吸附以及羟基的生成和转化是重要基元步骤,稀土氧化物独特的表面酸碱性能对该反应存在重要影响。与Al2O3、TiO2等氧化物载体相比,稀土离子具有更大的离子半径和更弱的路易斯酸性,表现出独特的CO2加氢性能。通过形貌控制和掺杂等手段综合调控表面氧空位浓度、金属载体相互作用以及表面酸性,是设计高效CO2加氢催化剂的有效策略。
1.3.4 甲烷重整反应
甲烷是天然气的主要成分,在石油工业中也常作为废气被燃烧或排放,其成本低廉且是一种比CO2效力更强的温室气体,将甲烷转化为高附加值产品具有巨大的经济效益和环境效益。目前,通过甲烷重整反应,将甲烷转化为合成气(CO+H2)并进一步转化为高附加值产物是其重要应用方向。甲烷重整的主要方法有甲烷蒸汽重整(MSR)和甲烷干重整(MDR),在这2个方法中常用的金属催化剂以贵金属和Ni为主,而稀土氧化物是其常见载体。
目前,对Ni/CeO2催化剂催化甲烷蒸汽重整的反应机理和构效关系已经有了相当充分的研究。Wu等利用DFT模拟方法深入研究了Ni/CeO2催化剂的结构和性能,发现在Ni/CeO2催化剂上,Ni的氧化态对反应催化性能有着重要影响,Ni4O/CeO2是其最佳氧化态形式,可以同时获得高活性和高稳定性[76]。Mierczynski等研究了载体对镍基催化剂催化甲烷含氧蒸汽重整的影响,发现CeO2∶La2O3为2∶1的载体对反应最为有利,表现出最高的活性和氢气选择性,这可归因于Ni与载体的强相互作用[77]。
相比于甲烷蒸汽重整,应用于甲烷干重整的催化剂更为丰富。由于在甲烷干重整反应中催化剂上易发生积炭,分散度更高,因此抗积炭性能更好的贵金属催化剂在甲烷干重整反应中得到广泛应用。例如,Zhang等研究了Pt/CeO2催化剂上金属载体相互作用对催化甲烷干重整反应的影响[78]。该研究发现:Pt(111)直接应用于甲烷干重整反应会发生严重积炭,导致催化性能不佳,但制备的Pt/CeO2催化剂则表现出优异的催化性能。进一步研究表明:在Pt/CeO2催化剂上,载体不仅可以通过金属载体相互作用调控Pt位点的电子结构,促进CH4解离,还可以直接参与CO2的吸附和解离。这一现象在本课题组的研究中也得到了证实[42]。Alabdullah等研究了不同载体上Rh纳米粒子的尺寸对甲烷和丙烷干重整性能的影响,发现Rh纳米粒子的尺寸与反应的催化活性存在紧密联系,Rh纳米粒子和载体界面对反应的关键步骤具有较高催化活性,较小的纳米粒子催化活性更高[79]。对传统Ni基催化剂的研究主要集中在提升抗积炭性能上。Jiang等研究了镍基甲烷干重整催化剂的耐硫和耐焦化性能,结果表明,向传统的Ni/CeO2催化剂中引入La或Zr可以提高其耐硫性能[80]。
甲烷重整反应的2种策略还可以联用,即甲烷混合重整。Karemore等研究了Ni-K/CeO2-Al2O3催化剂上甲烷混合重整的反应性能,其通过向催化剂中引入K来调控Ni的电子结构进而改善催化性能[81]。这一催化剂在800 ℃条件下取得了91.2%的CH4转化率,并可以通过调整进料中水蒸气和CO2的比例来调整产品合成气中CO和H2的比例。
甲烷重整反应的关键步骤是C—H键的活化,稀土氧化物本身不具备活化C—H键的能力,因此相比载体,负载于稀土氧化物上的金属位点更为重要。在设计甲烷重整反应的催化剂时,应当着重关注氧化物载体与负载金属之间的金属载体相互作用,尽可能保证活性金属的稳定分散。另外,甲烷重整反应气氛下容易生成非活性碳物种,进而导致催化剂焦化,因此耐焦化性能也是甲烷重整催化剂的重要评价标准。
1.3.5 其他应用
稀土氧化物材料除了上述几类经典的应用领域外,还有许多其他应用,例如合成氨反应、氨分解反应、燃料电池及光解水等。
Li等研究了不同稀土元素(La、Y和Pr)掺杂Ru/BaCeO3的合成氨反应催化性能[45]。在3 MPa、450 ℃条件下,2.5%的Ru/BaCe0.9La0.1O3-δ催化剂表现出最高氨气合成速率。La的引入有助于增加催化剂中氧空位的数量,并可以增加Ru表面的电子密度,从而提高催化活性。
Tan等通过简单的高温煅烧方法,将氧化钕(Nd2O3)引入管状g-C3N4材料形成复合催化剂,并将其用于光催化制氢反应和NO去除反应(图13)[18]。相比于原始的g-C3N4,该复合材料在光催化制氢反应中将H2生成速率提升了近10倍,在NO去除反应中将NO去除率提高了32%,表现出优异的催化性能。

图13 Nd2O3/g-C3N4复合材料的能级结构以及光催化机理Fig.13 Energy level structure and photocatalytic mechanism of Nd2O3/g-C3N4注:参考自文献[18]。
Li等制备了一系列负载在碳载体上的稀土氧化物电催化剂,并将其用于电催化ORR反应[35]。通过在NH3气氛下退火,可以向其中引入N原子掺杂以及氧空位,所获得的电催化剂具有优异的催化性能。
稀土氧化物催化剂有着非常广阔的应用场景,其一些应用实例总结于表2。目前,通过向金属或载体组分中掺入少量其他金属,进而调控金属载体相互作用,是一种行之有效的调控稀土氧化物负载型催化剂催化性能的方法。尽管经历了长时间的研究,但稀土氧化物催化剂领域仍有很多问题未能解决。例如,由于CeO2的变价性能,导致CeO2负载型催化剂的表面结构往往相当复杂,许多反应的活性位点和反应机理尚不明确。与CeO2结构和性能类似的PrO2催化剂尚未得到广泛关注和研究。此外,如何将稀土氧化物基催化剂的优异性能应用于电催化和光催化领域也尚未得出明确答案。这为稀土氧化物催化剂的发展提供了方向,可能成为未来关注的重点。

表2 稀土氧化物的应用实例Tab.2 Application examples of rare earth oxides
目前,稀土氧化物基催化剂已经得到相当充分的研究,对在纯稀土氧化物以及较为简单的两组分稀土氧化物负载型催化剂上进行的各种催化反应的反应机理和构效关系也有相当深入的认识,这极大地指导了稀土氧化物纳米催化剂的进一步发展。在这些已有研究的基础上,研究人员开发出了许多更为复杂的多组分催化剂,它们表现出比两组分催化剂更为优异的催化性能。然而,现有研究还难以清晰地阐明多组分催化剂中各种组分的详细作用。为了改进现有催化剂,仍然需要付出相当高的成本以完成试错。该领域的深入发展一方面需要借助各种高精度的先进表征手段,更加清晰地阐明多组分催化剂中各种组分的作用;另一方面也要借助人工智能等数据科学手段,在丰富的催化剂应用实例中找到半定量规律。
2 稀土合金纳米催化材料
合金化是一种提高金属催化剂性能的有效方法。稀土合金可以将稀土元素独特的电子结构和催化性能整合到传统的金属催化剂材料中,进而调整合金的能带结构和表面配位环境以提高催化性能。稀土合金具有较高的合金化焓,稳定性良好[90-91]。然而,与过渡金属及贵金属相比,稀土金属的还原电位非常低,稀土合金很难在通常的空气气氛或质子溶剂中进行合成。传统的物理合成方法虽然已经在磁性材料制备等领域得到了广泛应用,但物理方法或是只能获得块状金属锭,或是需要苛刻的合成条件,往往不适用于纳米催化剂的合成,限制了稀土合金的应用。当前,Pt-RE合金因其优异的稳定性和电催化活性备受关注,是研究最为热门的稀土合金,其他类型稀土合金的研究也在逐渐发展。
2.1 稀土合金的结构
根据组成元素的原子半径及其化学性质的差异,通常将合金分为固溶合金和金属间化合物2种,常用的稀土合金也大致分为这2类(图14)[92]。其中,金属间化合物是稀土合金催化剂的主要形式,稀土金属间化合物往往具有高度有序的原子排列,晶体结构独特。而在稀土固溶合金中,各种金属原子的排布是随机的,其晶体往往保持最密堆积形式,例如,Pt-RE合金保持了Pt金属的六方晶体结构[93]。
在实际设计和构造稀土合金催化剂时,往往会引入更多元素,包括非金属元素N、P、Si等,这大大提高了改性稀土合金的潜力。某些合金具有特殊的晶体结构,可以在催化过程中发挥独特功能。例如,Hosono课题组开发的LaScSi合金材料[94],具有金属间化合物和电子化合物的双重特性,其晶体结构中包含La4四面体和La2Sc4八面体2种空隙(图15),既可以容纳不同能量的电子,又可以可逆地存储氢原子。这一性质能够有效避免高H2分压下出现在Ru催化剂上的氢中毒现象,从而使Ru/LaScSi催化剂表现出相当高的合成氨催化活性,其合成氨反应活化能仅约为50 kJ/mol,明显低于各种氧化物基催化剂。

图14 稀土合金的结构Fig.14 Structures of RE-alloys注:参考自文献[92]。

图15 LaScSi和LaScSiH1.5的晶体结构及粉末XRD图谱Fig.15 Crystal structures and powder XRD patterns of LaScSi and LaScSiH1.5注:参考自文献[94]。
调节催化剂的电子结构是设计高效异相纳米合金催化剂的有效手段。通过稀土合金化,可以使电子在稀土元素和过渡金属元素之间重新分布,改变催化剂的电子结构,调节催化性能。由于稀土元素具有更负的还原电位,在稀土合金中往往会向过渡金属元素供给电子,导致过渡金属d带中心的移动和材料功函数的降低。Lim课题组研究了Pt-Y合金的d带中心与ORR反应电流密度之间的联系,发现电流密度与d带中心具有火山关系(图16),其中Pt70Y30处于火山口,解释了Pt3Y合金优异的ORR反应活性[95]。
2.2 稀土合金纳米催化剂的合成
稀土合金纳米催化剂的合成大体上有2种思路。一种是直接利用物理方法制备合金,而后设法将物理方法得到的材料处理成适合进行催化的纳米结构,例如,将块状合金熔铸成金属电极用于电催化反应。另一种是克服化学合成的困难,采用如非水相合成方法等新兴合成化学手段,直接合成稀土合金纳米颗粒,用于电催化反应。

图16 0.1 mol/L HClO4溶液中PtYx合金 催化ORR反应电流密度与d带中心之间的关系Fig.16 Kinetic current density (jk) for ORR at 0.9 V on the Pt-Y electrodes in a 0.1 mol/L HClO4 solution as a function of the measured d-band centers注:参考自文献[95]。
2.2.1 物理冶金法
物理冶金法通过非化学方法制备合金,是一种构造多晶稀土合金的有效策略。其常用方法是根据化学计量比对金属材料进行称重,放入感应炉或高温炉中,在惰性气氛保护下将材料熔融得到稀土合金。物理冶金法可以大批量制备稀土合金,适用于工业生产,获得的块体合金往往直接处理制成金属电极或者压碎制成粉末后使用。
Saccone课题组将化学计量的2种金属密封于钽坩埚中,在氩气气氛下感应加热的同时反复摇动坩埚,制备出一系列Pt-RE合金,并将其直接制成合金纽扣,抛光后用作硼氢化物燃料电池的阳极电极[96]。该方法也适用于更复杂合金体系的合成。Saccone课题组在最新的研究中用该方法合成了La2Pd3Ge5和Nd2Pd3Ge5合金材料,用来研究它们的电磁性质[97]。在Chorkendorff课题组的一项研究中,研究人员利用超高真空技术,将多晶Pt3Y和Pt3Sc合金置于1 273 K真空环境下退火,退火后Pt向合金表面偏析并形成纯Pt表面层,制备出的Pt-RE合金催化剂具有比单晶Pt更强的ORR反应活性[98]。
2.2.2 磁控溅射法
磁控溅射法使用Ar-N2中的等离子体混合气体在电场和交变磁场作用下产生的高能粒子轰击目标表面,使其原子脱离靶材并转移到衬底表面形成薄膜。通过选择靶材以及对电压、气压进行控制,可以制备出不同结构、密度、厚度、组成的稀土合金薄膜。磁控溅射法制备的稀土合金具有较为可控的结构和洁净的表面,非常适合用来合成模型催化剂。
Lim课题组利用磁控溅射法制备了一系列具有不同比例的Pt-M合金电极(M=Ti, Fe, Zr,La),其通过调整磁控溅射系统中的金属靶控制合金成分[99]。另外,前文提到的Lim课题组研究中所使用的一系列Pt-Y合金也是通过这种方法制备的[95]。
结合磁控溅射法和气体聚集技术,还能制备出尺寸均一的合金纳米颗粒。Chorkendorff课题组的另一项研究利用这一技术合成了负载在玻璃碳基质上的一系列不同尺寸和组成的PtxY纳米颗粒,并将其用作氧气电还原反应的模型催化剂[100]。
2.2.3 化学还原法
化学还原法可以在相对温和的条件下合成合金纳米颗粒。与物理方法相比,化学还原法制备的催化剂具有更大的比表面积,可以暴露更多的活性位点。然而,稀土金属过低的还原电势使其无法在水溶液或质子溶剂中合成,导致化学还原法曾长期受到限制,直到近几年才逐渐得到人们的关注。目前,研究人员已经开发出多种化学还原方法,成功合成了具有高比表面积的稀土合金材料。
Kanady等利用三乙基硼氢化物盐的低熔点,开发出熔融盐“溶剂”方法,用于合成Pt3Y合金(图17a)[101]。在上述合成过程中,不需要添加有机配体作为稳定剂,保证了催化剂表面的洁净;此外,还可以通过简单的洗涤将Pt3Y纳米颗粒转移到碳载体上,为制备电极提供了便利。类似地,Kobayashi等在LiCl-CaH2熔融体系中合成了三元金属合金RENi2Si2[102]。这表明熔融盐方法也适用于过渡金属-稀土合金的合成。
Li课题组通过另一种方法实现了Pt-RE合金的合成(图17b)[103]。该方法利用g-C3N4对金属离子较强的络合能力以及较差的热稳定性,首先将Pt和RE金属离子嵌入到g-C3N4网络中,再通过控制条件的H2热处理得到Pt-RE合金。该团队的最新研究对上述合成策略的机理进行了深入探究,阐明了Pt-RE合金形成的一系列化学过程,包括富氮前体的聚合、多孔RE2(CN2)3相的生成、碳载体上金属相的快速迁移以及高合金化热驱动的稀土金属还原等,为该合成策略的进一步优化和拓展奠定了基础[104]。
Ryoo等通过在沸石合成过程中加入Ga盐,继而用硝酸除去Ga原子的方法制备出带有表面骨架缺陷的中孔沸石;在该沸石上共浸渍Pt和La、Y,再在H2气氛下还原,制得三元催化剂[105]。上述研究发现:La、Y可以与Pt纳米颗粒以合金的形式共存,且它们的掺入使催化剂催化丙烷脱氢反应的寿命延长了10倍以上。该团队认为中孔沸石上的缺陷位点通过配位键来稳定单原子RE物种,这些单原子RE物种在后续的H2还原过程中与Pt结合生成合金。
2.2.4 电化学方法
在外电场的作用下,将RE离子和过渡金属/贵金属离子共沉淀在阴极表面也是合成RE合金的有效方法。与化学还原法相似,由于稀土金属过低的还原电势,电沉积时所用的电解质溶液必须具有足够宽的电化学窗口。离子液体或熔融盐是电沉积较为理想的电解质。例如,Gao等利用离子液体N-丁基-N-甲基吡咯烷鎓二氰胺作为电解质,在铜阴极上沉积了Ni-La合金膜[106]。Liu等通过在LiCl-KCl-SmCl3-CuCl2熔体中共还原Sm和Cu合成了Sm-Cu合金,并在成功得到Sm-Cu合金单晶产物的基础上,深入研究了共沉积过程中的晶体生长、相变以及不同沉积电势对Sm-Cu合金结构的影响[107]。

图17 熔融盐法制备Pt3Y合金(a)及g-C3N4诱导Pt-RE合金合成(b)示意图Fig.17 Schematic diagrams of preparation of Pt3Y alloy by molten salt method(a)and synthesis of Pt-RE alloy induced by g-C3N4(b)注:修改自文献[101]和[103]。
与自下而上的电沉积方法相反,阴极腐蚀方法可以自上而下地由大块稀土合金制备稀土合金纳米颗粒,从而有效提高金属原子的利用率。早在10年前,Yanson等就已经对Pt阴极腐蚀现象进行了较为详尽的研究,发现通过在阴极上施加交流电压,光滑的电极表面会在负电位下生成金属纳米颗粒,并在正电位下脱落[108]。Fichtner等利用这一策略,绕开复杂的自下而上合成Pt-RE合金的工艺,在不外加任何表面活性剂或化学还原剂的情况下,在KOH电解质中合成了PtxPr/C催化剂,该催化剂具有比商用Pt/C催化剂高约1.7倍的比质量活性[109]。
电化学方法也可以和通常的化学方法联用。例如,Gong等利用两步法合成了PtSmCo三元金属纳米颗粒,首先在离子液体中进行恒电位电化学沉积制备SmCo颗粒,再将SmCo颗粒与K2PtCl4进行置换,得到在SmCo颗粒上生长形成的PtSmCo三元金属颗粒[110]。
2.3 稀土合金的催化应用
稀土合金催化剂的应用主要集中在电催化领域,大部分情况下,稀土元素通过调控贵金属或过渡金属的电子结构来提高其电催化活性,因此稀土合金催化剂往往与其中的贵金属或过渡金属具有相同的应用场景。由于在电化学工作环境下,合金电极表面还原电势极低的稀土会逐渐溶解,并在表面形成富含另一种金属的富集层,因而目前认为稀土元素在反应物吸附等反应关键环节不直接发挥作用,其在合金中主要起到调节电子结构进而影响催化性能的作用。
2.3.1 氢析出反应
电解水生产氢气是解决化石能源危机的方案之一,具有广阔的发展前景。这一反应主要由氢析出反应(HER)和氧析出反应(OER)构成。目前,稀土合金催化剂已经被广泛应用于HER反应中。
HER反应具有相对简单的反应机理。在酸性电解质中,HER反应主要由2步组成[111]。在第一步中,催化剂表面的质子与电极上的电子结合,生成吸附态的H原子:
H(aq)++ e-= Hads。
(1)
随后的H2分子脱附有2种途径。一种是吸附态的H原子与溶液中的另一个质子反应,并与第二个电子结合,形成H2分子脱附:
Hads+ H(aq)++ e-= H2 (g)。
(2)
另一种是2个吸附态的H原子结合,生成H2分子脱附:
Hads+ Hads= H2 (g)。
(3)
由上述机理可以看出,催化剂表面H原子的吸附能对HER反应性能具有重要影响。Zhang等近期的研究结果也表明,在碱性电解质中,H吸附能是非常有效的HER性能描述符[112]。本质上,HER反应的2个步骤是相互竞争的,因此理想的HER催化剂应当具有适中的H原子吸附能(图18)[113]。

图18 HER反应的电流密度与纯金属上H原子吸附能 的DFT计算值之间的火山关系示意图Fig.18 Experimentally measured exchange curren for hydrogen evolution over different metal surfaces plotted as a function of the calculated hydrogen chemisorption energy per atom注:参考自文献[113]。j0的单位为A/cm2。
由图18可知,在HER反应的催化剂中,以Pt为主的贵金属占有重要地位,但贵金属催化剂高昂的成本限制了其工业应用。为了降低催化剂成本并进一步改善贵金属催化剂的催化性能,研究人员开发了一系列贵金属-稀土合金,并深入研究了它们的HER催化性能。早在2013年,Santos等就研究了Pt-RE合金(RE=Ce, Sm, Ho)在碱性电解质中催化HER反应的性能,发现稀土元素的引入提高了HER的催化活性,Pt-Sm合金表现出最好的催化性能[114]。
近期,非贵金属-稀土合金也得到了较多关注。Ghobrial等为了提高非晶态合金材料的电化学活性表面积,开发了Ni-Nb-Y合金体系[115]。这类合金在电化学反应条件下会原位生成异质结构,从而产生更大的电化学活性表面积。在碱性条件下,Ni81.3Nb6.3Y12.5多相催化剂表现出最好的HER催化活性。Zhu等在泡沫Ni上沉积了无定性Ni-Co-B-RE四元合金非晶态膜,并将其用于HER反应,其中Ni-Co-B-Gd/NF电极在碱性溶液中表现出优异的催化性能和电化学稳定性[116]。该研究表明,稀土元素与过渡金属之间的电子协同作用对其催化性能具有显著影响,Gd的添加还可以增加催化剂的表面积并提升电荷转移速率。Rosalbino等设计合成了三元Fe-Mo-RE合金,并将其用于催化HER反应,发现电极上H原子的溢流效应和不同合金相的协同效应可能对反应有着重要影响[117]。
对HER催化剂而言,H原子吸附能是最重要的催化性能影响因素。在HER合金催化剂的设计中,稀土元素通常用于调控合金的电子结构,进而调控H原子吸附能。通过改变稀土元素的种类以及掺入比例,可以在相当大的范围内灵活调控催化剂的H原子吸附能,这为HER催化剂的设计提供了有利条件,预期稀土合金催化剂在HER反应中将产生更为广泛的应用。
2.3.2 燃料电池
大部分燃料电池,如质子交换膜燃料电池(PEMFC)、乙醇燃料电池(DEFC)、锌空气电池(ZAB)等的阴极反应都是氧还原反应(ORR)。当前,Pt是低温ORR使用最为广泛的催化剂。早在2009年,就已经报道了Pt与Y、Sc合金化可以有效提升其ORR催化性能,并引起了研究人员的重视[90]。经过十几年的发展,ORR已经成为稀土合金的主要应用领域之一。

除前文提到的Lim课题组对Pt-RE合金催化剂ORR性能的研究外,还有许多稀土合金被应用于催化ORR反应的报道。Kim等研究了RE-Pd-Ir三元催化剂在ORR反应中的性能[119]。研究表明:合金催化剂d带中心的移动对ORR催化活性具有重要影响;稀土元素的掺入会造成Pd原子表面电子密度上升,使d带中心远离费米能级;稀土原子的几何效应对ORR反应性能也有影响。Gong等将Pt-Sm-Co三元金属纳米颗粒用于电催化ORR反应,取得了很好的反应活性,且该催化剂在酸性介质中催化ORR反应的稳定性优于商用Pt/C催化剂[110]。Brown等利用磁控溅射法合成了洁净的Pt3Y薄膜,并以此为基础深入研究了ORR过程中Pt3Y合金的表面结构,研究结果有助于新型高活性纳米催化剂的开发[120]。
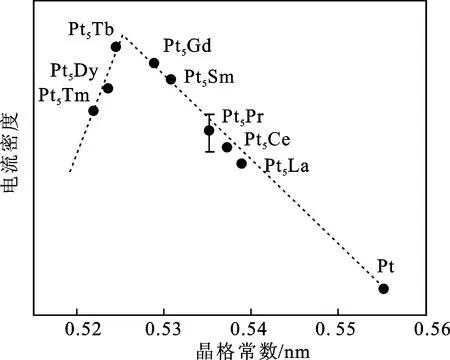
图19 Pt-RE合金催化剂ORR活性与六方 晶体晶格参数的火山关系Fig.19 Volcano curves obtained by plotting the kinetic current densities of Pt-Re polycrystalline electrodes versus their lattice parameters注:参考自文献[93]。

2.3.3 热催化反应
稀土合金材料由于具有独特的储氢性能,在许多与氢原子迁移密切相关的热催化反应中也有应用。
早在1996年,就有报道称LaNi5合金具有催化氨合成的活性[123]。近期,Ye等深入研究了LaNi5催化剂催化氨合成反应的机理,发现LaNi5合金在氨合成反应过程中会发生分解生成LaNi5核和LaN-Ni壳,表现出高活性和高稳定性[124]。该LaNi5纳米颗粒活性明显高于其他报道中Co和Ni基催化剂的活性,甚至可以与Ru基催化剂相媲美。
Ryoo等将负载在介孔沸石上的Pt-RE合金纳米粒子应用于丙烷脱氢(PDH)反应[105]。研究发现:与Pt催化剂相比,Pt3La和Pt3Y纳米粒子对PDH催化的多方面性能都有显著提升,包括反应活性、选择性和催化剂寿命等,而制备的Pt和RE未合金化的对照催化剂没有表现出相关性能的增强,初步表明La、Y与Pt纳米颗粒合金化对PDH反应催化性能的提升具有关键作用。
稀土合金催化剂的应用目前还比较局限,合金中稀土元素的作用主要是调节过渡金属或贵金属的电子结构,且稀土元素对合金电子结构的具体影响机制也有待进一步研究。对于HER、ORR等简单反应,稀土元素在合金材料中的作用已经较为清楚,借鉴已有研究方法,将研究范围拓展到如醇类氧化等复杂的电催化反应是该领域的一个重要发展方向。另外,稀土合金催化剂最主要的应用领域是各类电催化反应,但稀土元素在电催化条件下易于溶解到电解液中,因此实际反应中的催化剂与新制备的催化剂可能具有不同结构,当前研究尚不能清晰地阐明原位条件下催化剂的结构。要克服这一挑战,需要开发多种新型原位电化学表征手段,这也是稀土合金纳米催化剂研究的重大课题。
3 稀土助催化剂
稀土元素凭借其特殊的电子结构和配位性能,经常被掺入到其他催化剂中,以助催化剂的形式调控其他催化剂的催化性能。因此,稀土元素掺杂是一种设计和制备高效催化剂的有效策略。目前,稀土助催化剂的研究也得到了一定发展,例如Osazuwa等综述了稀土元素对CH4重整反应的影响[125],指出对于Ni基催化剂,稀土元素掺入会改变催化剂的还原性、吸附性能、晶体结构及形貌等多种性质,还能够通过参与反应决速步骤在CH4重整反应的反应机理及动力学中发挥关键作用。近期稀土元素作为助催化剂的应用实例详见表3。
总的来说,稀土可以通过调节氧空位浓度和表面酸碱性能、调控金属载体相互作用、诱导材料形貌变化等方式对催化材料的性能产生影响。目前,稀土元素助催化剂的研究主要集中在稀土元素对主体催化剂的调控上,对稀土元素在反应过程中的原子作用及其对催化活性位点的具体影响尚未形成明确认识。若能解决这一问题,则有望进一步拓展稀土助催化剂的应用范围,为高效催化剂设计提供新的灵感。

表3 稀土助催化剂应用实例Tab.3 Application examples of rare earth co-catalysts
4 稀土单原子催化剂
单原子催化剂(SAC)的概念最早出现于2011年[135],是指以原子分散的金属原子为活性中心的非均相催化剂。单原子催化剂因具有最高的原子利用率,展现出相当高的比活性,受到研究者的广泛关注。稀土元素的富电子轨道易于与各种配位原子生成配位键,有利于稳定稀土单原子催化剂,但稀土元素易于聚集,单独形成氧化物相,这使得稀土单原子催化剂难于合成。直到近两年,稀土单原子催化剂才逐渐走进研究人员的视野,属于新兴催化剂体系,报道较少。
Zhu等利用硬模板法成功构建了原子分散的Ce位点,并嵌入分层的氮掺杂碳催化剂中,Ce以CeN4O6的高配位数构型存在(图20)[136]。该催化剂利用SiO2球形模板,经掺杂、酸洗和气体迁移3步合成,实现了较高的活性Ce位点密度。作者通过球差校正的电子显微镜、X射线吸收光谱等表征手段探明了催化剂中Ce位点的配位环境,并将这一催化剂应用于质子交换膜燃料电池中,表现出优异的ORR催化活性,其半波电位为0.862 V,功率密度高达0.525 W/cm2。

图20 CeN4O6的配位结构和制备方法示意图Fig.20 Schematic diagram of coordination structure and preparation method of CeN4O6注:修改自文献[136]。
Ji等开发出一种新颖的原子束缚和配位策略,实现了碳氮纳米管上负载的单原子Er合成[137]。该研究首先将三聚氰胺海绵在含有尿素和硝酸铒的溶液中浸泡并剧烈搅拌,使其均匀填充前驱体溶液;随后用液氮冷却并冻干至完全除去水分,在N2气氛下退火,得到锚定在碳氮纳米管上的Er单原子(图21)。该策略利用低温下尿素和三聚氰胺的配位作用限制了Er3+随机迁移,实现了Er3+的原子级分散。通过调整Er前驱体的用量,可以得到Er负载量高达20%的单原子催化剂。将这一材料用于光催化CO2还原反应,一系列表征及DFT计算表明,原子分散的Er原子对高效的光催化CO2还原性能有杰出贡献。

A. Er1/CN-NT催化剂合成示意图;B. 低负载量Er1/CN-NT的HAADF-STEM图像和相应元素分布图;C. 低负载量Er1/CN-NT的AC HAADF-STEM图像;D. 图C虚线框区域的放大图像;E. 高负载量Er1/CN-NT的HAADF-STEM图像和相应元素分布图;F. 高负载量Er1/CN-NT的AC HAADF-STEM图像;G. 图F虚线框区域放大图像。
Liu等利用固相反应方法合成了Y1/NC和Sc1/NC催化剂[138]。该团队利用ZIF-8为结构模版,通过向ZIF-8中掺杂RE,再在高温下热解来得到稳定的Y1/NC和Sc1/NC。结构表征结果表明:这些材料中的RE原子通过与C、N原子配位而被稳定下来;不同于过渡金属单原子催化剂中常见的M-N4配位结构,Y原子与6个氮或碳原子配位。Y1/NC和Sc1/NC催化剂在氮气还原(NRR)和二氧化碳还原(CO2RR)反应中都表现出了优异的催化性能。
目前,通过合理设计配位结构及利用先进合成手段限制RE原子迁移和聚集,已经能够有效地合成稀土单原子催化剂。在稀土单原子催化剂中,稀土原子作为活性中心,表现出独特的催化活性。当前,对稀土单原子的研究和应用尚处于起步阶段,还没有总结出成熟的合成方案,对其表现出的独特催化性能也缺乏原子级别的清楚认识,这些挑战可能会成为未来稀土单原子催化剂的研究方向。
5 总结与展望
本文从稀土异相纳米催化剂的各种存在形式出发,重点介绍了稀土氧化物、稀土合金、稀土助催化剂、稀土单原子催化剂4种稀土异相纳米催化剂,综述了4种催化剂的结构、控制合成方法以及它们在催化应用中的研究进展。这4种稀土异相纳米催化剂的研究进展以及面临的主要挑战总结于表4。

表4 稀土异相纳米催化剂的研究进展及主要挑战Tab.4 Research advances and main challenges of heterogeneous rare earth nanocatalysts
随着稀土异相纳米催化剂研究的不断发展,研究人员也面临着新的挑战。总结而言,对稀土异相纳米催化剂的研究主要面临两项挑战:其一,通常的催化反应条件非常复杂,多组分纳米异相催化剂的结构也相当复杂,对催化剂在反应条件下的原位表征较为困难,对已经报道的、具有优异催化性能的诸多催化剂的催化反应机理和活性位点附近原子级的结构认识还不够清晰。其二,多组分催化剂中各组分对催化剂结构的影响较为复杂,且催化剂结构与催化性能之间的关系常常是非线性的,这导致尽管已经报道了大量比传统两组分负载型催化剂催化活性更好的多组分催化剂,但尚未总结出其半定量规律或模型,新催化剂的设计与开发相当困难,需要研究人员付出大量努力。
稀土异相纳米催化剂的发展方向可以基于上述两项主要挑战进行展望。针对第一项挑战,随着合成化学的发展,具有明确结构的纳米催化剂合成变得更加容易,对催化性能存在影响的各种结构因素,如暴露的晶面、负载元素的粒径、多元催化剂中各组分的分布等,都可以得到更有效的控制,这将有利于催化剂构效关系的研究。另外,原位表征技术的发展也为稀土异相纳米催化剂研究提供了更为丰富的手段。在已有成果的基础上,一方面可以借助合成化学的最新进展,制备结构更为可控的模型催化剂;另一方面也需要研究人员积极设计与开发适合于具体反应环境的原位测试方案。
针对第二项挑战,稀土元素彼此间较为接近的化学性质为这一问题提供了可能的解决途径。通过相邻稀土元素对稀土催化剂的掺杂,可以实现催化剂结构和性能在较小范围内的连续变化,结合已有研究手段,在晶格常数、缺陷密度、材料能带结构等结构数据与各关键中间物种的吸附能、过渡态能垒等催化性能数据之间建立定量或半定量关系。以此为基础,将有机会建立适用范围更广泛的模型,为催化剂的设计提供指导。此外,以人工智能和大数据科学为基础的最新数据科学方法为这一领域提供了全新的研究工具,但其在催化剂研究中的应用目前还存在诸多限制,例如,不同实验室得到的催化数据同源性较差,影响催化剂性能的变量过多等。随着高通量实验方法的发展和机器人实验员性能的进步,上述问题有望逐渐克服,可以期待数据科学方法将来成为稀土异相催化剂研究的有力工具。
