制备方法对FeMnCeOx催化剂(超)低温NH3-SCR性能的影响
2022-03-18孙敬方蔡彦迪余雅昕安冬琦田笑丛邹伟欣
孙敬方,蔡彦迪,余雅昕,安冬琦, 田笑丛,邹伟欣*,董 林,,*
(1 南京大学 现代分析中心,江苏省机动车尾气污染控制重点实验室,江苏 南京 210093; 2 南京大学 环境学院,江苏 南京 210093; 3 南京大学 化学化工学院,教育部介观化学重点实验室,江苏 南京210093; 4 南京大学 化学国家级实验教学示范中心,江苏 南京 210093)
近年来,我国经济、社会的高速发展导致对煤炭等化石能源的需求量逐年增加,该形势下伴随而生的是一系列如酸雨、雾霾等严重的环境问题[1]。氮氧化物(NOx)作为大气环境污染的重要源头之一,其排放来源可分为两类:一类是固定污染源,主要包括电力、钢铁、水泥、玻璃窑炉等行业排放的工业烟气;另一类是移动污染源,主要为汽油车、柴油车、船舶等移动交通运输工具的尾气[2]。虽然自2013年《大气污染防治行动计划》(《大气十条》) 发布和实施以来,我国在国家和地方层面采取了一系列大气污染防治措施,使大气环境污染问题得到了明显改善,但不少地区和城市仍然面临着空气质量不达标的巨大压力。此外,近年来臭氧问题的凸显及“碳达峰、碳中和”要求的提出,也对我国大气环境治理提出了更高要求。
《大气十条》发布后,我国对燃煤电厂污染的治理取得了显著成绩,其氮氧化物排放量基本达到了国家规定的超低排放标准。2019年4月,生态环境部等五部委又联合发布了《关于推进实施钢铁行业超低排放的意见》(环大气[2019]35号),以实现全流程、全过程环境管理为目标,推动实施钢铁行业的超低排放,这使得钢铁行业成为全国第二个实现超低排放的行业。可以预见的是,电力和钢铁行业的超低排放改造必将促进相应排放控制技术的发展,进而带动整个非电行业氮氧化物处理技术的革新。NH3选择性催化还原(NH3-SCR)技术作为燃煤电厂常用的烟气脱硝技术,在非电行业中同样具有广阔的应用空间[3-4]。但是,电力行业中使用的NH3-SCR商用催化剂V2O3-WO3(MoO3)/TiO2的反应温度窗口(300~400 ℃)与非电行业的烟气温度范围(80~280 ℃)不匹配,传统烟气升温使用催化剂又会带来过高能耗,进而限制了NH3-SCR技术在非电行业的应用[5]。因此,亟需开发出适用于非电行业烟气实际工况的(超)低温NH3-SCR脱硝催化剂[6-7]。
在诸多金属氧化物催化剂中,锰基催化剂由于具有丰富的表面活性氧物种和多变的金属离子价态,在低温NH3-SCR反应中展现出巨大潜力[8]。但是,锰基催化剂存在高温N2选择性差、工作温度窗口窄和易中毒(SO2和H2O、碱/碱土金属、重金属)等问题,限制了其工业化应用[9]。通过改进制备方法、元素掺杂改性、完善预处理过程等方式提升锰基催化剂脱硝性能的研究一直备受关注[10-12]。
本研究拟利用Fe2O3和CeO2优异的低温性能及抗硫性能,设计合成FeMnCeOx催化剂,并比较不同制备方法(溶胶-凝胶法、共沉淀法)对FeMnCeOx(超)低温NH3-SCR性能的影响,探究影响催化剂低温活性的关键因素,以期为合成高效低温抗硫型NH3-SCR催化剂提供理论指导。
1 材料与方法
1.1 催化剂制备
1.1.1 溶胶-凝胶法
先取一定量的2 mol/L柠檬酸溶液于20 mL水中,再加入计算量的Mn(CH3COO)2·4H2O、Fe(NO3)3·9H2O和Ce(NO3)3·6H2O,室温下剧烈搅拌并在120 ℃条件下干燥过夜。将所得棕黑色样品研磨均匀,置于500 ℃马弗炉中焙烧3 h。
保证所有样品中柠檬酸和金属离子的物质的量比为1,Ce的质量分数固定为12.5%。将n(Fe)∶n(Mn)=1∶1的样品简记为FeMnCeOx(S),n(Fe)∶n(Mn)=2∶3的样品简记为Fe2Mn3CeOx(S)。
1.1.2 共沉淀法
准确称取一定量的Mn(CH3COO)2·4H2O、Fe(NO3)3·9H2O和Ce(NO3)3·6H2O溶于20 mL水中,室温下磁力搅拌2 h。在上述溶液中逐滴加入NH3·H2O,控制溶液的最终pH值为10。抽滤沉淀液体后得到块状固体,放入120 ℃烘箱中干燥过夜。将干燥样品研磨均匀,并在空气气氛下500 ℃焙烧3 h,得到FeMnCeOx(C)催化剂。催化剂中Ce质量分数固定为12.5%,将n(Fe)∶n(Mn)=1∶1和2∶3的样品分别简记为FeMnCeOx(C)和Fe2Mn3CeOx(C)。
1.2 催化剂表征
1.2.1 XRD表征
样品的XRD表征在X-衍射仪(X′pert Pro,Philips)上完成。仪器采用Cu靶Kα辐射(0.154 08 nm波长)和Ni滤波片,工作电流为40 mA,工作电压为40 kV。
1.2.2 N2吸脱附实验
采用ASAP-2020物理吸附仪(Micromeritics)进行N2吸脱附测试,表征样品的比表面积、孔容和孔径。
1.2.3 H2-程序升温还原(H2-TPR)
H2-TPR在实验室自组装的多功能化学吸脱附分析仪上完成,仪器采用TCD检测器。称取10 mg催化剂样品置于U型管中,N2气氛下200 ℃吹扫30 min,降至室温后切换成体积分数7%的H2/Ar混合气体,以10 ℃/min的升温速率由室温程序升温至850 ℃,同时收集数据。
1.2.4 NH3-程序升温脱附(NH3-TPD)
NH3-TPD在实验室自组装的多功能化学吸脱附分析仪上完成。称取50 mg样品置于U型管中,在300 ℃、He气氛中吹扫60 min(10 mL/min);降至室温后切换成NH3直至吸附饱和;再次用He气吹扫30 min,去除物理吸附的NH3物种;最后在He气氛中(10 mL/min)以10 ℃/min的升温速率程序升温至650 ℃。记录NH3物种的脱附信号。
1.2.5 原位漫反射红外实验(insituDRIFTS)
insituDRIFTS在配备有漫反射附件HARRICK的Nicolet 5700型原位傅里叶变换红外光谱仪上进行(ZnSe窗片),仪器使用MCT检测器,图谱采用Kubelka-Munk函数进行表达。实验过程中,待测样品首先在400 ℃、纯N2气氛中吹扫30 min,降温至120 ℃并采集背景信号;然后切换为NO+O2气体直至吸附饱和,再在N2气氛下(50 mL/min)吹扫30 min;最后切换为NH3气体。采集随时间变化的红外图谱,得到先通入NO+O2气体后通入NH3气体的实验结果。
类似地,对于先通入NH3气体后通入NO+O2气体的实验,先通入NH3气体直至吸附饱和,再用N2气体吹扫,最后切换为NO+O2气体。采集随时间变化的红外图谱。
1.3 催化剂性能评价
催化剂样品的活性评价实验在自搭建的固定床连续流动反应器上进行。准确称取200 mg样品(60~80目)至石英管中,在250 ℃、N2气氛中吹扫30 min,降至室温后切换成反应气氛直至吸附饱和。反应气体中NH3、NO和O2的体积分数分别为0.05%、0.05%和5%,平衡气为Ar气,反应空速为60 000 mL/(g·h)。随后以10 ℃/min的升温速率升温至设定温度,反应达到稳态后进行数据采集。得到的产物使用在线Nicolet IS10 型色谱进行检测分析,催化剂的NO转化率采用以下公式进行计算:

其中[NO]in和[NO]out分别代表NO气体的入口浓度和出口浓度。
2 结果与讨论
2.1 催化剂的性能评价
2.1.1 催化剂的脱硝性能
图1为采用2种制备方法制备的FeMnCeOx催化剂样品的NO转化率。可以看出,无论采用何种Fe/Mn配比,溶胶-凝胶法制备的锰基催化剂样品的催化活性都明显高于共沉淀法制备的催化剂样品。Fe2Mn3CeOx(S)和FeMnCeOx(S)均可在80 ℃时达到90%的NO转化率,并在80~150 ℃温度区间内维持稳定。此前汪楷迪等[13]所报道的Fe2Mn2Ce6Ox-WO3/TiO2催化剂在130 ℃时的NO脱硝率仅为43.9%;其在使用NiO和CuO共同改性Fe2Mn2Ce6Ox-WO3/TiO2后,改性催化剂在相同条件下的脱硝率仅提升至73.9%。这充分说明本研究所制备的催化剂在脱硝性能方面具有优越性。与此同时,无论采用溶胶-凝胶法还是共沉淀法,Fe/Mn配比为2∶3样品的催化活性均优于配比为1∶1的样品。因此,本研究后续选取Fe/Mn配比为2∶3的样品系统考察不同方法所制备催化剂活性存在差异的主要原因。
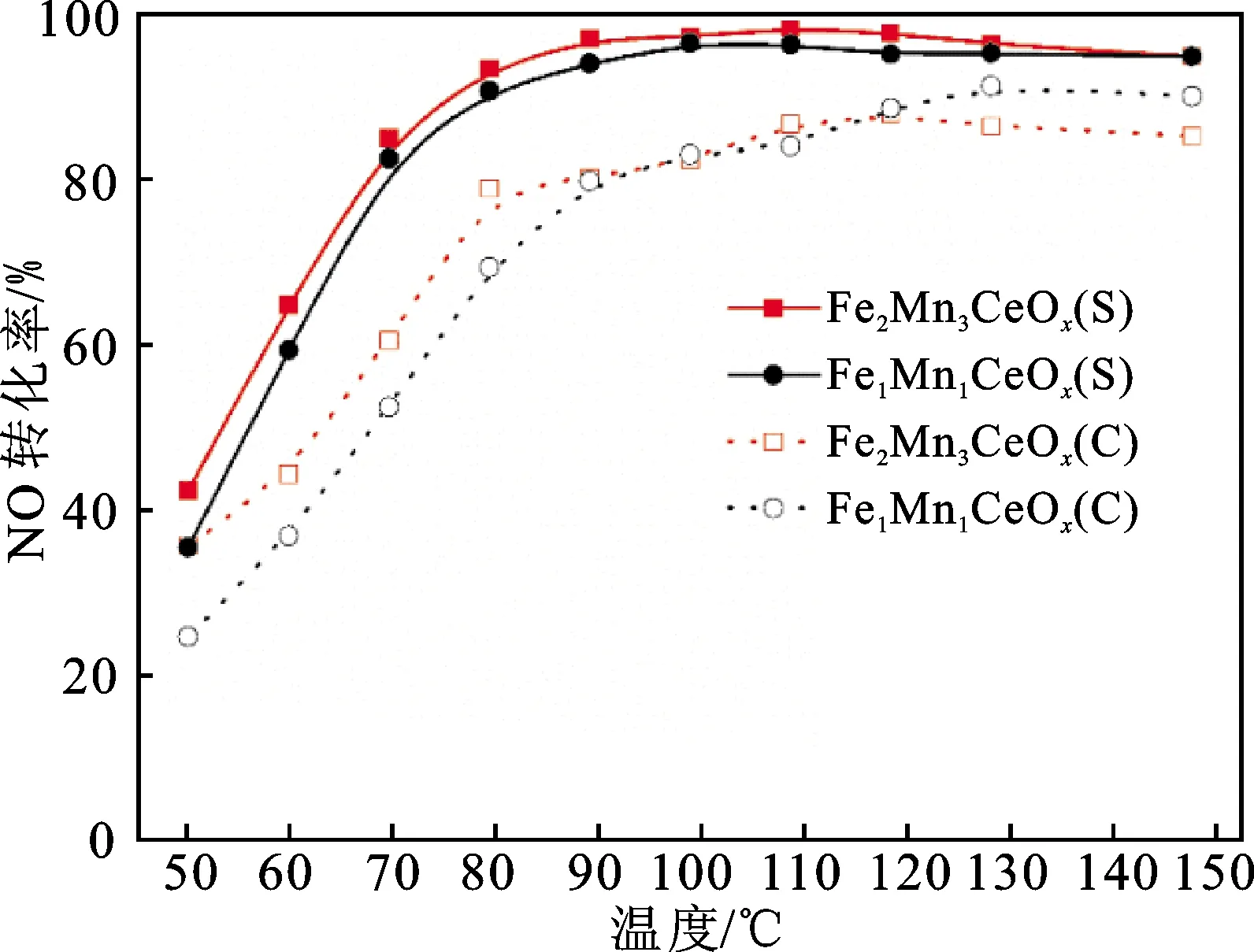
图1 催化剂样品的NO转化率Fig.1 NO conversion rates of the catalyst samples
2.1.2 催化剂的抗硫性能
在实际工况反应中,虽然会采取除尘、脱硫等工艺对烟气进行前处理,但在脱硝反应的烟气中仍不可避免会有少量SO2残存,而残存的SO2对催化剂具有毒害作用。对于锰基催化剂而言,抗硫性能是直接影响其催化性能的关键因素。基于此,本研究考察了不同制备方法对催化剂样品抗硫性能的影响。由图2可知,在150 ℃的反应条件下,向反应气氛中通入体积分数为0.01%的SO2后,Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)的NO转化率具有明显差异。Fe2Mn3CeOx(S)催化剂在通入SO2后催化性能没有发生显著变化,仍可在反应11 h时保持95%以上的NO转化率。但Fe2Mn3CeOx(C)催化剂的NO转化率受SO2气氛影响较大,具体表现为在通入SO2约3 h后,该催化剂的催化活性开始持续下降,其在抗硫10 h时的NO转化率已下降至70%左右,11 h时更是跌落至50%左右。以上结果表明,采用溶胶-凝胶法制备的催化剂样品不仅具有优异的脱硝性能,而且具备良好的抗SO2性能。
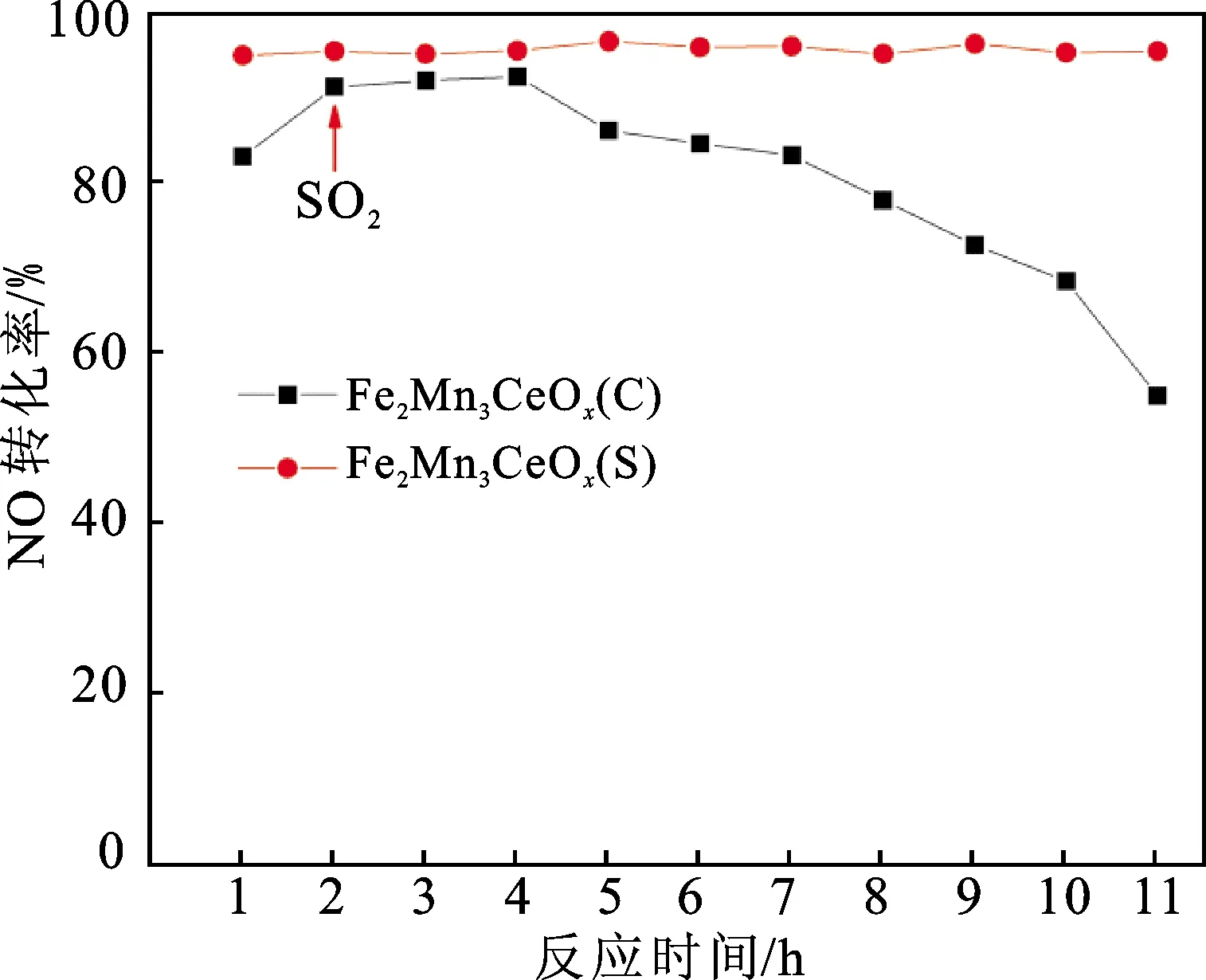
图2 150 ℃条件下Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C) 的抗SO2中毒性能Fig.2 SO2 resistance of Fe2Mn3CeOx(S) and Fe2Mn3CeOx(C) at 150 ℃
2.2 XRD和BET分析
为探究2种方法所制备催化剂的脱硝反应活性存在差异的根本原因,对催化剂的结构特征进行了XRD和BET表征。XRD表征结果揭示了2种催化剂的晶体结构(图3),可以看出,Fe2Mn3CeOx(S)催化剂主要表现出2种晶体结构衍射峰,即Fe3Mn3O8的晶相衍射峰(PDF-ICDD 75-0034)和立方萤石结构CeO2(PDF-ICDD 34-0394)的晶相衍射峰[14-15]。相比之下,Fe2Mn3CeOx(C)催化剂的晶相衍射峰强度明显减弱,仅能观测到位于30°左右的宽包,其中隐约可以看出归属于Fe3Mn3O8和CeO2物种的晶相峰主峰。由此推测采用溶胶-凝胶法制备的催化剂样品脱硝性能优异的原因可能与其较好的晶相结构有关。

图3 Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)的XRD结果Fig.3 XRD profiles of Fe2Mn3CeOx(S) and Fe2Mn3CeOx(C)
对于多相催化反应,特别是气固相催化反应,催化材料的比表面积和孔结构会影响反应物分子的吸脱附性质及其与催化剂表面的接触,进而影响催化活性。为深入探究织构性质对催化剂活性的影响规律,对2种方法制备的锰基催化剂样品进行了N2吸脱附和孔径分布分析,结果如图4所示。由图4a可以看出,Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)样品都表现出典型的Ⅳ型N2吸脱附等温线,这意味着2种催化剂的孔结构均为介孔结构[16]。由图4b可以看出,Fe2Mn3CeOx(S)样品的孔径分布更为均匀,主要集中在约5 nm;而Fe2Mn3CeOx(C)样品的孔径分布较为分散,孔径尺寸明显增大,在8~12 nm都有分布。

V代表孔体积,D代表孔径。图4 Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)的N2吸脱附等温线(a)及孔径分布(b)Fig.4 N2 adsorption-desorption isotherms (a) and pore size distributions (b) of Fe2Mn3CeOx(S) and Fe2Mn3CeOx(C)
2种催化材料的比表面积、孔容和平均孔径在表1中列出。可以看到,Fe2Mn3CeOx(S)的3项结构参数均明显低于Fe2Mn3CeOx(C)。这可能是因为采用溶胶-凝胶法制备的催化剂样品孔道容易收缩和塌陷,致使催化剂的比表面积和孔容变小[17]。由于Fe2Mn3CeOx(S)样品的NO转化率明显高于Fe2Mn3CeOx(C)样品,推测催化剂的织构性质不是造成其脱硝性能差异的关键原因。
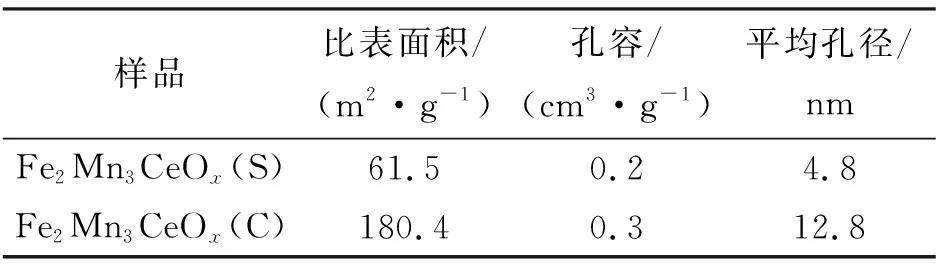
表1 Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)的结构参数Tab.1 Structural parameters of Fe2Mn3CeOx(S) and Fe2Mn3CeOx(C)
2.3 氧化还原性能(H2-TPR)
在NH3-SCR反应中,适宜的氧化还原性能对反应物分子的活化至关重要。为评价2种方法所制备催化剂的氧化还原能力,对样品进行了H2-TPR测试,结果如图5所示。溶胶-凝胶法制备的Fe2Mn3CeOx(S)样品出现有3个明显的氧化还原峰,分别位于287 ℃、445 ℃和740 ℃附近。其中,位于287 ℃附近的还原峰归属于铁物种Fe2O3到Fe3O4的还原;位于445 ℃左右的宽包归属于Mn3O4到MnO的还原及Fe3O4到FeO的还原[18];位于740 ℃的还原峰主要归属于催化剂中CeO2物种的还原[19]。

图5 催化剂样品的H2-TPR结果Fig.5 H2-TPR profiles of the catalyst samples
与Fe2Mn3CeOx(S)相比,Fe2Mn3CeOx(C)的还原峰位置显著向高温方向偏移,同时位于200 ℃左右的还原峰强度大幅减弱,只出现了位于395 ℃和628 ℃左右的2个还原峰,说明该催化剂的氧化还原能力较Fe2Mn3CeOx(S)明显减弱。一般而言,催化剂的氧化还原性能直接影响NH3-SCR反应物分子NO的活化,特别是在低温阶段,因此溶胶-凝胶法制备的Fe2Mn3CeOx(S)催化剂具有较好的低温氧化还原能力可能是其表现出较高脱硝反应活性的重要原因。
2.4 表面酸性(NH3-TPD)
在NH3-TPD检测中,催化剂的NH3吸附能力被直观地表现为催化剂表面酸性位点NH3分子吸附随温度的变化情况[20],结果如图6所示。可以发现,Fe2Mn3CeOx(S)样品在450 ℃左右出现有1个较大的脱附峰;相比之下,Fe2Mn3CeOx(C)样品的脱附峰强度明显减弱,只在250 ℃和580 ℃附近出现有2个微弱的小峰。结果表明,Fe2Mn3CeOx(S)样品的表面酸量明显高于Fe2Mn3CeOx(C)样品,该结果与催化剂的NO转化率结果一致。
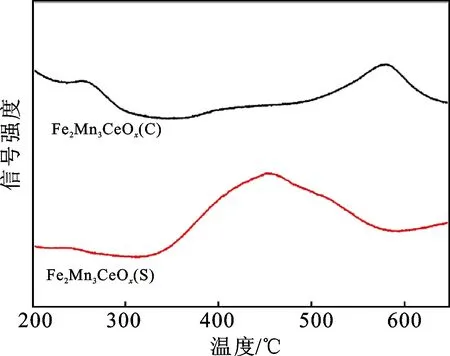
图6 催化剂样品的NH3-TPD结果Fig.6 NH3-TPD profiles of the catalyst samples
2.5 表面元素分析(XPS)
利用XPS对不同制备方法所得催化剂的表面元素化学状态进行表征,结果如图7所示。图7a中的Fe 2p图谱可以观察到Fe 2p3/2(710.0~711.0 eV)和Fe 2p1/2(723.5~724.5 eV)2个信号峰[21]。图7b中有2个Mn 2p信号峰分别归属于Mn4+(642.9 eV)和Mn3+(641.8 eV)。由此可以看出,Fe2Mn3CeOx(S)催化剂表面具有更高Mn4+/Mnn+比,而Mn4+在低温SCR反应中能够促进NO转化为NO2,所以含有更高Mn4+/Mnn+比的Fe2Mn3CeOx(S)催化剂的低温SCR活性更佳[22]。图7c中的Ce 3d图谱显示Fe2Mn3CeOx(S)催化剂的Ce3+含量高于Fe2Mn3CeOx(C)催化剂,而有研究表明催化剂中Ce3+含量较高有利于NH3-SCR催化性能的提升[23]。图7d中的O 1s图谱显示Fe2Mn3CeOx(S)催化剂含有更多的表面活性氧Oα。据文献报道,表面活性氧Oα比晶格氧Oβ具有更强的流动性和氧化能力,在SCR反应中扮演着重要角色,更有利于反应进行[24]。由此可知,Fe2Mn3CeOx(S)更多的表面活性氧含量也是其具有较好低温脱硝性能的促进因素。

图7 催化剂样品的XPS谱图Fig.7 XPS spectra of the catalyst samples
2.6 反应机理(in situ DRIFTS)
为揭示Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)催化剂的NH3-SCR反应路径,通过原位红外实验探究了其反应机理(图8)。

图8 Fe2Mn3CeOx(S)与Fe2Mn3CeOx(C)的原位红外光谱Fig.8 In situ DRIFTS spectra of Fe2Mn3CeOx(S) and Fe2Mn3CeOx(C)
首先将Fe2Mn3CeOx(S)暴露在120 ℃、NH3气氛中直至吸附饱和,随后通入NO+O2气体,随着时间的迁移观察催化剂表面的变化(图8a)。当NH3吸附饱和后,Fe2Mn3CeOx(S)出现了在1 173、1 196、1 275 cm-1处吸附在L酸位点的NH3信号峰以及1 445 cm-1处吸附在B酸位点的NH3信号峰[25-26]。随后再通入NO+O2气体,Fe2Mn3CeOx(S)表面吸附的NH3信号峰逐渐消失,并出现了1 567、1 243 cm-1处的双齿硝酸盐信号峰、1 275 cm-1处的单齿硝酸盐信号峰以及1 616 cm-1处的桥式硝酸盐信号峰[27-28],证明吸附在Fe2Mn3CeOx(S)表面的NH3可以与气态的NO+O2发生反应。对Fe2Mn3CeOx(S)进行先NO+O2吸附饱和后通入NH3的实验(图8b),发现催化剂表面吸附的硝酸盐物种在通入NH3后没有明显变化,说明吸附的硝酸盐物种不与气态的NH3发生反应。
由图8c、8d可以看出,Fe2Mn3CeOx(C)催化剂的NH3-SCR反应路径与Fe2Mn3CeOx(S)催化剂相似。但根据各气体分子信号峰的出现及消失速度可知,Fe2Mn3CeOx(C)催化剂表面的反应过程进行较慢,这可能是其催化活性低于Fe2Mn3CeOx(S)催化剂的原因。
综上所述,Fe2Mn3CeOx(S)和Fe2Mn3CeOx(C)催化剂表面的NH3-SCR反应是通过Eley-Rideal(E-R)机理进行的。对于典型的E-R反应机理,NH3首先吸附在L酸位点上生成-NH2等中间产物,这些中间产物继而与气相NO/NO2反应生成N2和H2O。具体反应过程如下:
NH3+*→NH2-*+H,
(1)
NH2-*+NO→N2+H2O。
(2)
Fe2Mn3CeOx(S)催化剂的表面酸性强,可以获得更多的NH3吸附量,进而明显促进上式的进行,因此具有更高的NO转化率。
3 结语
通过溶胶-凝胶法和共沉淀法制备了FeMnCeOx催化剂,考察了不同制备方法对催化剂低温脱硝性能的影响,并通过织构性质、氧化还原性、表面酸性及表面物种化学态表征探究了催化剂的具体反应机制。相比于共沉淀法制备的FeMnCeOx(C),采用溶胶-凝胶法制得的FeMnCeOx(S)催化剂表现出更优异的催化性能,其在80~150 ℃温度范围内获得了超过90%的NO转化率,且在120 ℃、体积分数为0.01%的SO2气氛中活性稳定,耐硫性能良好。理化特性表征结果表明,Fe2Mn3CeOx(S)具有更佳的氧化还原性能、更强的表面酸性及更丰富的利于低温SCR反应的表面物种,有利于反应物分子的吸附活化;Fe2Mn3CeOx(S)的NH3-SCR反应遵循E-R反应机理,催化剂表面吸附的NH3越多越有利于反应发生。
