水相铜催化α-酮酸与邻氨基苯硫酚合成苯并噻唑化合物
2022-03-16赖胤龙周丽珍陈梓深杜煜龙李秋燕谢汉非严绍熙
赖胤龙, 周丽珍,2, 陈梓深, 杜煜龙, 李秋燕, 谢汉非, 张 琦, 严绍熙*
(1.韶关学院 化学与土木工程学院, 广东 韶关 512005;2.广东省韶关生态环境监测中心站, 广东韶关 512005)
苯并噻唑化合物是一类重要的杂环化合物,具有良好的生物活性,是许多药物天然产物和功能分子的重要的母体和先导骨架,被广泛应用于医药、农药、工程材料等领域[1-4]。目前,苯并噻唑衍生物的经典合成方法是以邻氨基苯硫酚与羰基化合物(如醛、羧酸、羧酸衍生物等)为原料进行缩合反应[5-7]。然而,这类方法大多涉及高温、强酸和高沸点有机溶剂等苛刻条件。因此,发展一种原料简单易得、反应条件温和的苯并噻唑化合物的合成方法具有重要的现实意义。
酮酸在脱羧的反应中具有自身独特的特质,可以提供一种理想的碳合成子[8]。近十几年,酮酸作为脱羧酰化反应中酰化剂的来源已被广泛研究[9-12]。然而,利用α-酮酸和邻氨基苯硫酚通过脱羧环化方法合成苯并噻唑化合物的方法还比较稀少[13-18]。2014年,Lei 课题组首次报道了Ru催化剂在可见光条件下,利用α-酮酸和邻氨基苯硫酚脱羧环化合成了少数实例的苯并噻唑化合物(Scheme 1a)[13]。随后,Huang小组发展了一种电化学条件下,在添加酸和碱的共同作用下,恒电流电解α-酮酸和邻氨基苯硫酚生成了苯并噻唑化合物(Scheme 1b)[14]。2016 年,Laha课题组实现了在K2S2O8氧化剂作用下,α-酮酸钾盐和邻氨基苯硫酚的脱羧环化生成苯并噻唑类化合物的研究(Scheme 1c)[15]。Penteado小组分别报道了利用特殊的铌催化剂[16]和化学当量的Na2S2O5[17]合成苯并噻唑化合物的反应(Scheme 1d, 1e)。最近,Sharma课题组发现了在氧化剂H2O2存在下,利用蓝光的LED照射α-酮酸和邻氨基苯硫酚制备苯并噻唑的方法(Scheme 1f)[18]。由上可知,利用α-酮酸和邻氨基硫酚为原料合成苯并噻唑化合物的方法取得了较大进步,但仍存在需要使用有机溶剂、昂贵的过渡金属催化剂、特殊的转换装置、过量氧化剂等缺点。

Scheme 1
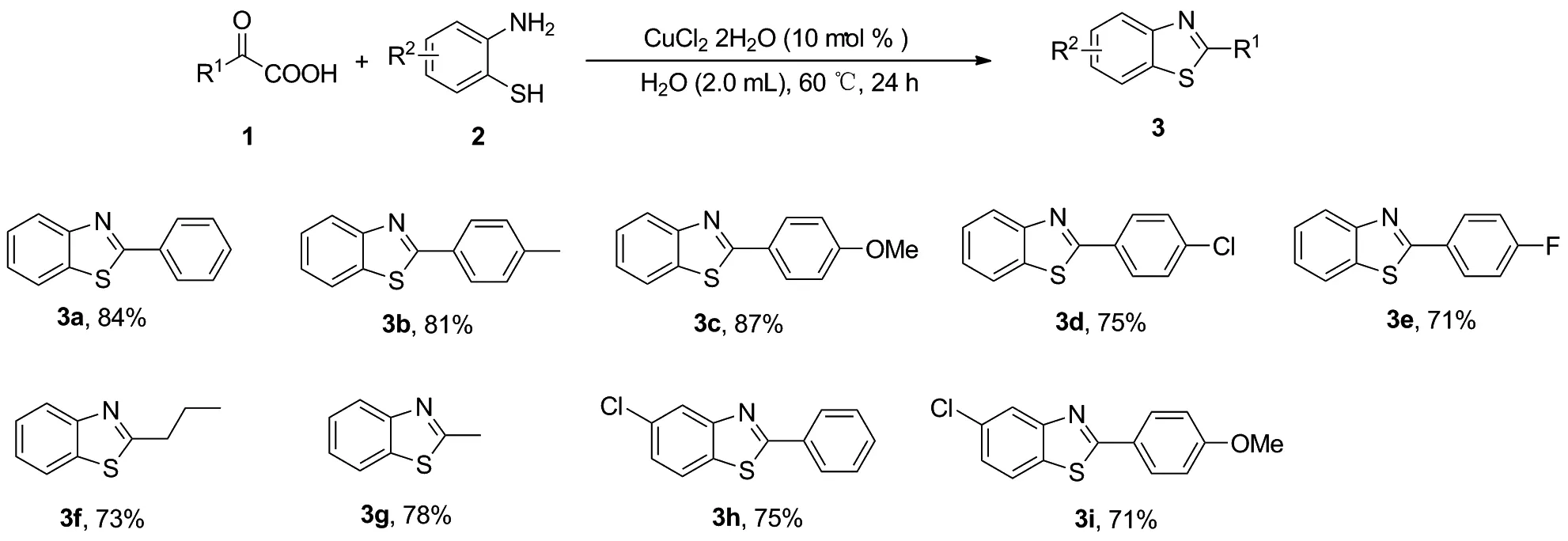
Scheme 2
水是一种廉价、安全且无污染的绿色溶剂,克服了大多有机溶剂的缺点,且可以产生新的反应性,因此水相反应越来越受到人们的关注[19-21]。此外,铜催化剂及其配体的易得性和低毒性使得铜催化羧酸氧化脱羧反应成为绿色化学的重要发展方向[22-24]。如能寻找一种水相条件下的铜催化α-酮酸和邻氨基苯硫酚合成苯并噻唑化合物的方法将具有重要的意义。近年来本课题组在水相和金属催化反应方面取得了较多成果[25-27],本文在此基础上,尝试以水为溶剂,廉价的CuCl2·2H2O为催化剂,在温和条件下催化α-酮酸和2-氨基硫酚合成苯并噻唑类化合物的反应。
1 实验部分
1.1 仪器与试剂
Bruker 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标)。
所用试剂均为分析纯。
1.2 3a~3i的合成(以3a为例)
在30 mL的带塞反应管中,依次加入苯乙酮酸1a(0.5 mmol),邻氨基苯硫酚2a(1.0 mmol), CuCl2·2H2O(0.05 mmol)和H2O(2 mL),于60 ℃反应24 h。反应结束后,加入饱和食盐水淬灭反应,并用乙酸乙酯(3×10 mL)萃取,合并有机相,干燥、过滤、滤液浓缩、残余物经硅胶柱层析纯化得化合物3a。
用类似的方法合成3b~3i。
2-苯基苯并噻唑(3a)[18]:白色固体,收率84%;1H NMR(400 MHz, CDCl3)δ: 8.12~8.09(m, 3H), 7.91(d,J=8.0 Hz, 1H), 7.52~7.49(m, 4H), 7.41~7.38(m, 1H);13C NMR(100 MHz, CDCl3)δ: 168.2, 154.3, 135.2, 133.8, 131.1, 129.2, 127.7, 126.5, 125.4, 123.4, 121.8。
2-(对甲苯基)苯并噻唑(3b)[18]:白色固体,收率81%;1H NMR(400 MHz, CDCl3)δ: 8.07(d,J=8.0 Hz, 1H), 7.99(d,J=4.0 Hz, 2H), 7.89(d,J=8.0 Hz, 1H), 7.51~7.47(m, 1H), 7.39~7.36(m, 1H), 7.31~7.29(m, 2H), 2.43(s, 3H);13C NMR(100 MHz, CDCl3)δ: 168.4, 154.3, 141.6, 135.1, 131.1, 129.9, 127.6, 126.4, 125.1, 123.2, 121.7, 21.7。
2-(4-甲氧基苯基)苯并噻唑(3c)[18]:白色固体,收率87%;1H NMR(400 MHz, CDCl3)δ: 8.05~8.02(m, 3H), 7.87(d,J=8.0 Hz, 1H), 7.47(t,J=8.0 Hz, 1H), 7.35(t,J=8.0 Hz, 1H), 6.99(d,J=8.0 Hz, 2H), 3.87(s, 3H);13C NMR(100 MHz, CDCl3)δ: 168.0, 162.0, 154.3, 135.0, 129.2, 126.5, 126.4, 124.9, 122.9, 121.7, 114.5, 55.6。
2-(4-氯苯基)苯并噻唑(3d)[18]:白色固体,收率75%;1H NMR(400 MHz, CDCl3)δ: 8.06(d,J=8.0 Hz, 1H), 8.02~7.99(m, 2H), 7.89(d,J=8.0 Hz, 1H), 7.52~7.44(m, 3H), 7.41~7.38(m, 1H);13C NMR(100 MHz, CDCl3)δ: 166.8, 154.2, 137.2, 135.2, 132.3, 129.4, 128.9, 126.6, 125.6, 123.5, 121.8。
2-(4-氟苯基)苯并噻唑(3e)[18]:白色固体,收率71%;1H NMR(400 MHz, CDCl3)δ: 8.10~8.06(m, 3H), 7.89(d,J=4.0 Hz, 1H), 7.50(t,J=8.0 Hz, 1H), 7.40~7.37(m, 1H), 7.20~7.16(m, 2H);13C NMR(100 MHz, CDCl3)δ: 166.90, 164.62(d,JC-F=200.0 Hz), 154.27, 135.22, 131.12(d,JC-F=3.0 Hz), 129.69(d,JC-F=7.0 Hz), 126.58, 125.42, 123.36, 121.78, 116.21(d,JC-F=18.0Hz)。
2-丙基苯并噻唑(3f)[14]:黄色固体,收率73%;1H NMR(400 MHz, CDCl3)δ: 7.97(d,J=8.0 Hz, 1H), 7.82(d,J=8.0 Hz, 1H), 7.43(t,J=4.0 Hz, 1H), 7.33(t,J=8.0 Hz, 1H), 3.08(t,J=8.0 Hz, 2H), 1.95~1.86(m, 2H), 1.05(t,J=8.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 172.3, 153.4, 135.3, 126.0, 124.7, 122.6, 121.6, 36.4, 23.2, 13.8。
2-甲基苯并噻唑(3g)[18]:棕色固体,收率78%;1H NMR(400 MHz, CDCl3)δ: 7.94(d,J=8.0 Hz, 1H), 7.81(d,J=8.0 Hz, 1H), 7.45~7.32(m, 2H), 2.83(s, 3H);13C NMR(100 MHz, CDCl3)δ: 167.2, 153.6, 135.8, 126.1, 124.9, 122.6, 121.6, 20.3。
5-氯-2-苯基苯并噻唑(3h)[18]:白色固体,收率75%;1H NMR(400 MHz, CDCl3)δ: 8.07~8.05(m, 3H), 7.78(d,J=8.0 Hz, 1H), 7.50~7.47(m, 3H), 7.36~7.34(m, 1H);13C NMR(100 MHz, CDCl3)δ: 170.1, 155.1, 133.5, 133.4, 132.5, 131.5, 129.2, 127.8, 125.8, 123.2, 122.4。
5-氯-2-(4-甲氧基苯基)苯并噻唑(3i)[18]: 白色固体,收率71%;1H NMR(400 MHz, CDCl3)δ: 8.00~7.98(m, 3H), 7.75(d, J=8.0 Hz, 1H), 7.32~7.30(m, 1H), 7.00~6.97(m, 2H), 3.87(s, 3H);13C NMR(100 MHz, CDCl3)δ: 169.9, 162.4, 155.2, 133.3, 132.3, 129.4, 126.2, 125.33, 122.8, 122.3, 114.6, 55.6。
1 结果与讨论
1.1 条件优化
以3a的合成为例,对反应条件进行了优化(表1)。由表1可知,当反应温度60℃,在H2O(2 mL)溶剂中反应24 h,发现可获得收率18%的目标产物3a(Entry 1)。受此启发,对不同的过渡金属进行了考察,发现CoCl2·6H2O、MnCl2·4H2O、FeCl3·6H2O和CuCl2·2H2O等过渡金属对反应均有较好的促进作用,其中CuCl2·2H2O的催化效率最好(Entries 2~5)。在此基础上对不同的铜化合物进行了筛选,结果显示,不论是其他类型的二价铜,还是一价铜,均能够以较高的收率得到目标产物,其中CuCl2·2H2O的效果最佳(Entries 6~11)。随后,进行了溶剂的优化,如乙腈(CH3CN)、1,4-二氧六环(1,4-dioxane)和N,N-二甲基甲酰胺(DMA)。结果显示H2O的效果最优(Entries 12~14)。接下来研究了反应温度的影响,结果表明60℃的反应收率最高(Entries 15~16)。最后研究了反应时间对反应的影响,发现反应时间对收率影响不明显(Entries 17~18)。
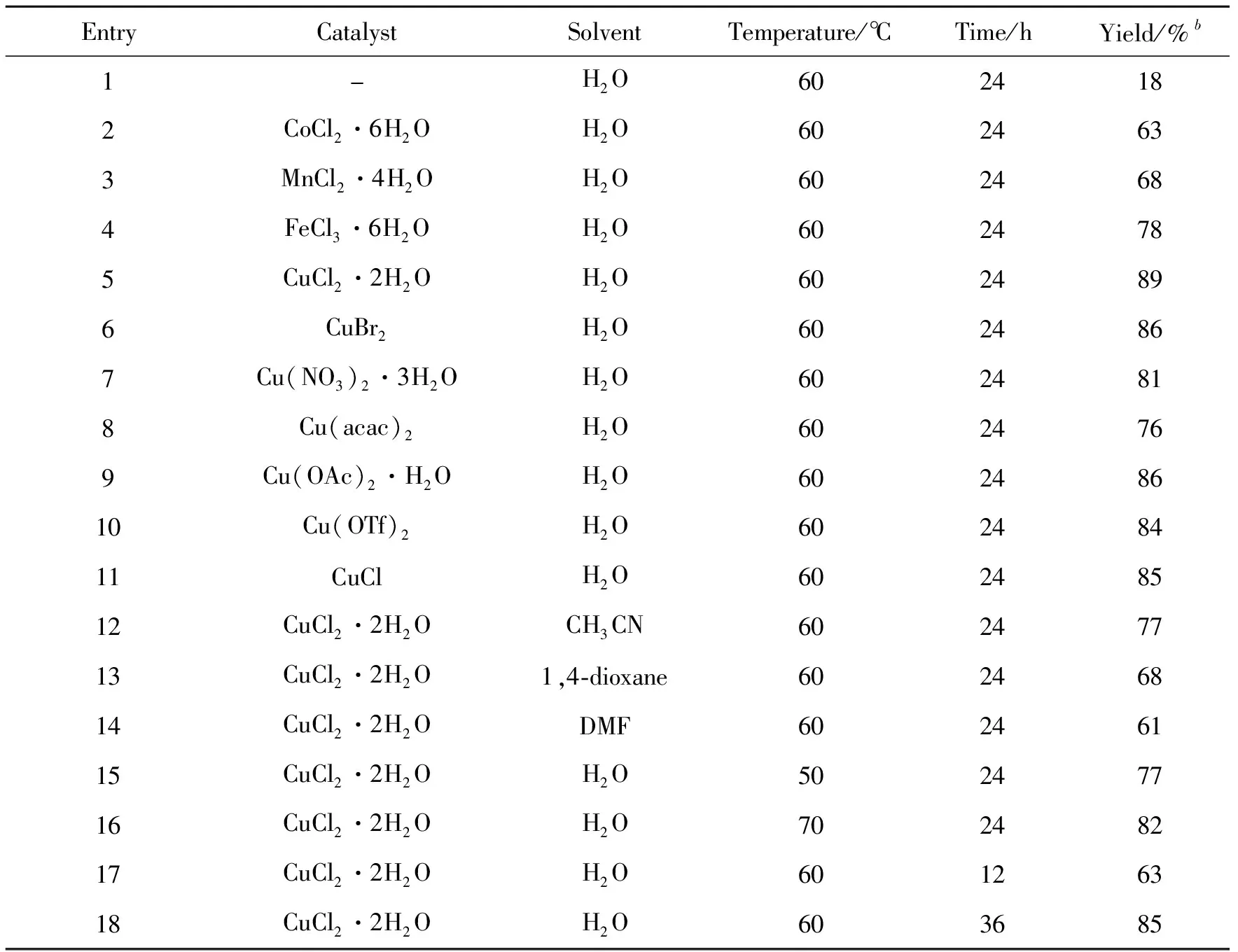
表1 反应条件的优化
2.2 底物拓展
确定了最优反应条件后,对底物普适性进行了考察,结果见Scheme 2。考察了化合物1芳环Ar 不同取代基对反应的影响,当在苯环上引入给电子取代基(如4-甲基或4-甲氧基)时,反应分别获得81%和87%收率的目标产物3b和3c;当在苯环上引入吸电子取代基时,如4-氯和4-氟时,亦能获得较好的收率,得到化合物3d~3e;接着发现烷基的酮酸效果也较好,可以获得73%和78%收率的3f和3g化合物;最后对化合物2的芳环进行考察,发现具有氯等基团时,反应也可以得到较好的产率的目标产物3h和3i。
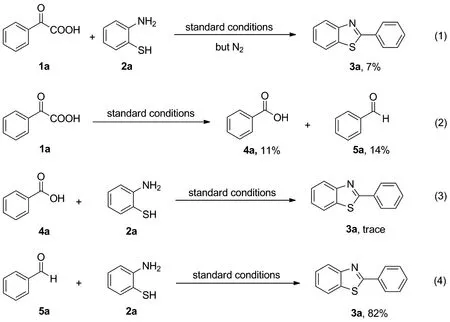
Scheme 3
2.3 控制性实验
为了探究反应的机理,进行了控制性实验,如Scheme 3所示。首先把标准条件改为氮气环境下反应,发现只能获得7%的收率,这表明了氧气对反应起到了重要的作用(eqs 1)。接着只有反应物1a在标准条件下反应,获得了11%的苯甲酸4a和14%的苯甲醛5a,这表明苯甲酸4a和苯甲醛5a可能是反应的中间体(eqs 2)。随后,以苯甲酸4a和邻氨基苯硫酚2a为原料,在最优反应条件下反应只能得到trace的目标产物,这表明苯甲酸4a可能不是反应中间体(eqs 3);最后以苯甲醛5a和邻氨基苯硫酚2a为反应底物,在最优反应条件下得到了82%的产物3a,这表明苯甲醛5a可能是反应的关键中间体(eqs 4)。
2.4 反应机理
通过控制性实验和相关文献的报告[5-7,22-24],提出了可能的反应机理(Scheme 4):苯乙酮酸1a在二价铜和氧气的共同作用下经过氧化脱羧生成中间体苯甲醛5a,苯甲醛5a快速与邻氨基苯硫酚2a进行缩合反应生成中间体6a和亚胺中间体7a,随后中间体6a和7a经过分子内的环化生成中间体产物8a,最后在二价铜和氧气的协助下中间体产物8a经过氧化去氢生成目标产物3a。
开发了一种新颖的铜催化水相的α-酮酸与邻氨基苯硫酚合成苯并噻唑化合物的方法。反应条件温和,具有较好的普适性,不同电性取代的α-酮酸与邻氨基苯硫酚均能顺利的获得较高产率的相应产物。该方法为合成苯并噻唑衍生物提供了一种简便可行的途径。
