AGL基因突变致糖原累积症Ⅲ型1例报告并文献复习
2022-03-15杨书婷王春林方燕兰孔元梅王卡娜许照洁
杨书婷 王春林 方燕兰 孔元梅 王卡娜 许照洁
糖原累积症(Glycogen storage disease,GSD)是一种较为罕见的遗传代谢性疾病,由糖原合成或降解过程中酶的缺乏引起,在存活新生儿中的发病率约为1/20 000~1/43 000[1]。糖原合成和分解代谢过程至少需要8种酶,这些酶缺陷导致至少16种不同类型的GSD亚型,其中GSD Ⅰa、Ⅲa和Ⅸa型主要受累器官为肝脏,分别由G6PC、AGL和PHKA2基因突变引起,分别对应葡萄糖6-磷酸酶、糖原脱支酶和磷酸化酶激酶的缺乏[2]。
目前报道最多的GSD亚型是GSD Ⅲ型,GSD Ⅲ型是由糖原脱支酶缺乏引起的糖原代谢障碍性疾病,该酶是糖原分解的关键酶,具有寡聚-(1,4→1,4)-葡萄糖转移酶和淀粉-1,6-葡萄糖苷酶双重催化功能,由AGL基因编码[3]。当AGL基因发生突变,糖原脱支酶合成障碍,糖原支链不能被分解,大量异常糖链蓄积于患者的肝脏和(或)骨骼肌、心肌中,临床表现为肝脏肿大、空腹低血糖、生长迟缓、进行性肌无力、心肌病等,其中以肝脏肿大最常见。随着年龄的增长,患儿低血糖和肝脏肿大有缓解趋势,而肌病表现越来越重,另有部分患儿可能合并肝腺瘤及肝硬化[4-5]。本文回顾分析2020年12月30日浙江大学医学院附属第一医院收治的1例AGL基因突变致GSD Ⅲ型患儿的临床资料,现报道如下。患儿 男,2岁3个月,因“发热3 d,咳嗽2 d”于2020年12月30日入院。患儿系孕1产1,孕足月剖宫产,出生体质量3 500 g,无窒息抢救史,无惊厥史,否认毒物接触及特殊药物使用史。自4月龄时腹膨隆明显,吃奶、精神反应无异常,家长未予重视,1岁时身高82 cm,较同龄儿偏高,1岁后身高增长速度变慢。目前2岁余,夜间睡前、凌晨醒后仍需进食。有一同父异母哥哥,14岁,体健,父亲家族中有肥胖家族史(具体不详),母亲体健,否认遗传病家族史。入院查体:体温38.5℃,心率136次/min,呼吸34次/min,血压 95/66 mmHg(1 mmHg=0.133 kPa),身高88.2 cm,体重15 kg,神志清楚,精神尚可,无特殊面容,咽充血,扁桃体Ⅱ°,双肺呼吸音粗,可闻及痰鸣音,心音中,未闻及心脏杂音,腹膨隆,触软,肝肋下8 cm,质软,脾肋下5 cm,双下肢无水肿,神经系统查体阴性。生化检测示转氨酶及肌酸激酶明显升高、脂质代谢异常(TC和TG升高,HDL降低、极低密度脂蛋白升高),复查生化指标(空腹)结果见表1。铜蓝蛋白、TORCH检查、凝血功能、EB病毒系列、铁蛋白、肝炎病毒抗原抗体测定、血气分析+乳酸、葡萄糖脑苷脂酶活性均未见明显异常,骨髓涂片细胞学检查未见泡沫细胞,心脏彩超无明显异常,肝胆胰脾彩超提示肝脾肿大(右肝斜径10.7 cm,肋下8 cm,肝实质回声分布均匀,脾肋下5.1 cm,脾实质回声均匀)。胸腹部CT提示双肺纹理增粗、模糊,边缘不清,肝外形肿大,肝实质未见异常密度灶。肾上腺素刺激试验[6]结果见表2,患儿空腹状态下经肾上腺素刺激后血糖较前上升<2.5 mmol/L,餐后经肾上腺素刺激后血糖上升>2.5 mmol/L。
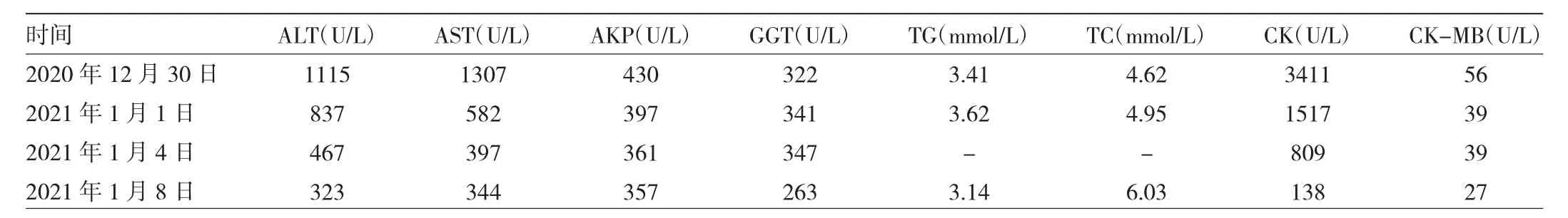
表1 患儿住院期间生化动态指标

表2 肾上腺素刺激试验
患儿自幼腹部膨隆明显,肝脾肿大,为明确病因,经家长同意后,采集患儿及其父母静脉血,从中提取基因组DNA行全外显子组测序,采用IDT The xGen Exome Research Panel v2.0全外显子捕获芯片捕获并测序,目标序列测序覆盖度约99%,结果发现:患儿AGL基因存在2处变异,分别为父源性c.3239(exon24)_c.3240(exon24)insT(致病证据为PVS1+PM2)及母源性c.4559(exon34)G>C(致病证据为PM1+PM2+PM3+PP3)。根据美国医学遗传学会遗传变异分类标准与指南(2015年),2处变异均判定为高致病可能,为新发现的变异,见图1-2。

图1 患儿及其父母AGL基因测序图[患儿携带AGL:c.3239(exon24)_c.3240(exon24)insT(p.C1080Cfs*29),患儿父亲相同位点杂合变异,患儿母亲相同位点未见突变]
该患儿有肝脏肿大、生长迟缓表现,辅助检查示肝酶明显升高、脂代谢异常、肝脾肿大,肾上腺素刺激试验阳性,结合基因检测结果,确诊为GSD Ⅲ型,住院期间予还原型谷胱甘肽及复方甘草酸苷静脉滴注护肝治疗,因有发热、咳嗽表现,X线胸片检查提示双肺纹理增粗,病原学检查提示呼吸道合胞病毒感染,故予雾化、氨溴索静脉滴注止咳化痰等治疗,经治疗后患儿肝酶进行性下降,体温正常,咳嗽好转,予出院后生玉米淀粉喂养、继续口服护肝药物,门诊定期复查。
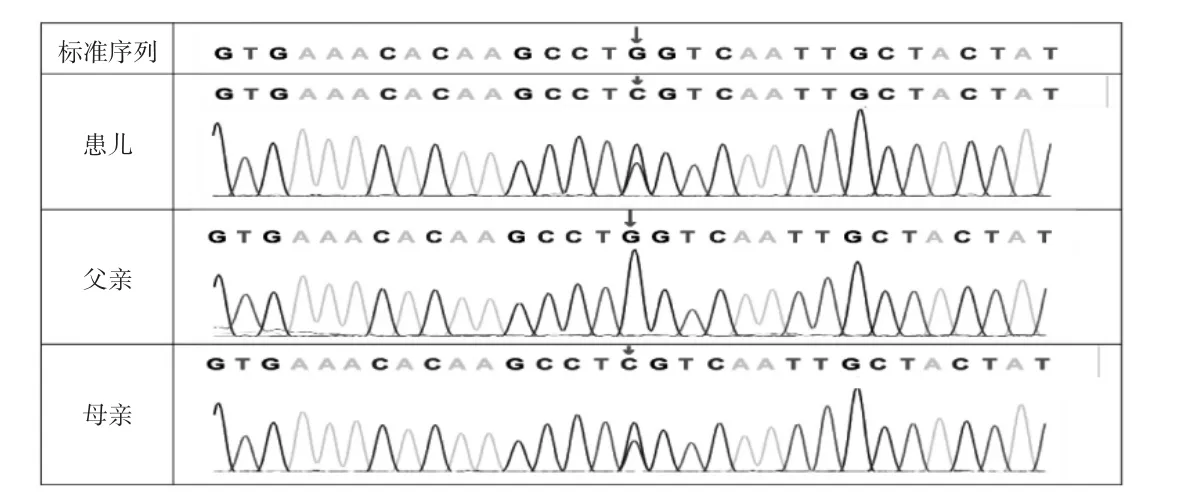
图2 患儿及其父母AGL基因测序图[患儿携带AGL:c.4559(exon34)G>C(p.W1520S),患儿母亲相同位点杂合变异,患儿父亲相同位点未见突变]
讨论GSD是由先天性酶缺陷造成糖原代谢异常的一组代谢性疾病,根据致病基因、酶缺陷的不同分为多种亚型,根据主要受累器官的不同又可分为肝GSD和肌GSD[7]。在GSD中,多种类型有相似的临床表现,如肝肿大、转氨酶升高、低血糖、高脂血症、尿酮阳性、生长迟缓等,仅依靠临床体征及实验室检查不能准确区分,基因检测为GSD的确诊、分型、判断预后提供了可靠依据,尤其是对于有GSD先证者的家系,强烈建议进行家系分析和基因检测[8]。在各型GSD中,除Ⅸa型为X连锁隐性遗传外,其他类型GSD均为常染色体隐性遗传[9-10]。
GSD Ⅲ型由AGL基因突变所致,AGL基因位于染色体1p21上,由35个外显子组成,基因组DNA长约85 kb,编码约7 kb的mRNA,通过选择性剪接产生至少2个启动子区、至少6种亚型,亚型1(启动子区1)为主要亚型,在肝脏、肌肉和卵巢中发挥作用,编码的糖原脱支酶含1 532个氨基酸残基,分子量约170 KD,亚型 2、3、4(启动子区 2)为肌肉特异性亚型,仅在肌肉细胞中发挥作用[7,11]。因此,根据糖原脱支酶催化活性及受累器官的不同,GSD Ⅲ型可分为a、b、c、d 4 种类型,以Ⅲa最常见,约占所有GSD Ⅲ型的80%,该型患儿除肝脏受累外,还有肌肉(如心肌、骨骼肌)受累;Ⅲb型约占Ⅲ型的15%,患儿仅肝脏受累;Ⅲc型患儿的葡萄糖苷酶活性缺乏,葡萄糖转移酶活性正常,仅肌肉受累;Ⅲd型患儿葡萄糖苷酶活性正常,葡萄糖转移酶缺乏,累及肌肉和肝脏,但Ⅲc和Ⅲd两型极为罕见[12-13]。
Perveen等[14]研究指出目前全球已发现258个AGL基因变异,不同种族的AGL基因变异具有异质性。Lu等[15]报道了我国41个家系,共43例GSD Ⅲ型患儿的基因测序结果,共检测到51个基因突变,包括剪接位点突变、小缺失突变、无义突变、重复突变、错义突变、复合缺失突变、插入突变等多种突变类型,发现最常见的突变方式为c.1735+1G>T,且该研究发现提前终止密码子与血清肌酸激酶升高有显著相关性。因此GSD Ⅲ型患者的基因型与表型之间可能存在一定相关性,有待进一步研究。
有文献指出,随着年龄的增长,GSD Ⅲ型患儿低血糖和肝脏增大情况有所缓解,但可能出现肝硬化、肝腺瘤、肝细胞癌等肝脏并发症,且部分患儿肌病表现有加重趋势,最常见的为心肌受累、骨骼肌受累,其中心肌病在心脏彩超上与原发性肥厚性心肌病相似,因此建议对不明原因的心室肥厚患儿需考虑GSD可能[12]。
GSD Ⅲ型患儿的早期治疗,可实行多样化饮食管理,如多餐饮食、生玉米淀粉和持续滴胃喂养等,达到预防低血糖发作的目的。对于反复持续的TG升高,可使用烟酸、贝特类等降TG药物,对反复持续的胆固醇浓度升高,可使用他汀类药物,对严重的高脂血症可选择进行血浆置换,但也有文献指出GSD Ⅲ型患儿使用他汀类降脂药会诱发或恶化肌病,故应谨慎应用他汀类降脂药[12,16-17];对发展为终末期肝病患者,可考虑行肝移植术。本例患儿经住院治疗后转氨酶较前下降,遂予出院,建议生玉米淀粉喂养,定期门诊复查。
综上所述,本例患儿以肝脏肿大、转氨酶升高、高脂血症为突出临床表现,空腹经肾上腺素刺激后血糖较前上升<2.5 mmol/L,餐后经肾上腺素刺激后血糖上升>2.5 mmol/L,经基因检测确诊为AGL基因复合杂合突变所致的GSD Ⅲ型,其突变位点此前未检索到相关报道,为新发现突变位点。临床上对于肝脏肿大、肝酶高、脂代谢异常的婴幼儿需要警惕GSD可能,对疑似GSD患儿建议尽早进行基因检测,达到早期明确诊断、精准治疗。
