Ge,Al掺杂锰硅化和物Mn4Si7的第一性原理计算
2022-03-04高振江丁庆昌
丁 璨, 胡 兴, 高振江, 丁庆昌
(三峡大学 电气与新能源学院, 宜昌 443002 )
1 引 言
热电材料是一种能够直接将电能和热能进行相互转化的半导体功能材料[1].在众多的热电材料中,金属硅化物就有着很好的应用前景,金属硅化物就是金属元素与硅元素结合所形成的化合物,如CrSi2,MoSi2,NbSi2等.因为其熔点高(大部分都在1500 ℃以上),电阻率低,硬度高,且含量丰富,利用率高,成本低等优点,所以被广泛应用于超大规模集成电路中,可用作金属栅,欧姆接触等,而其中高锰硅化合物(Higher manganese silicide,简称HMS)[2]MnSi1.7因其具有良好的光电性能,与传统的太阳能电池板相比,HMS的化学性质更加稳定,使用年限更加长久,并且对环境无污染,是目前已知金属硅化物中最良好的p型半导体材料[3].在块状晶体中,HMS有四种相结构,即Mn4Si7,Mn11Si19,Mn15Si26,Mn27Si47[4],它是一种抗氧化性好,电导率高,含量丰富,对环境无污染的良好半导体材料[5].
近些年来,随着人们对材料结构和性能的深入研究,人们开始利用分子模拟软件来构建分子模型,将不同的元素进行掺杂,进而可以得到在目前现实生活中无法发现或难以得到的物质,从而来预测新材料的结构和性质.关于对MnSi1.7的研究越来越多,Gottlied等人进行实验发现40k时Mn4Si7晶体出现微小的流动性,会产生了很小的有效磁矩[6].Zhou等人通过感应融化热压制备含有Ge添加剂的多晶高锰硅化物,从而提高了HMS的品质因数[7].Aoyama等人通过用Ge对Mn4Si7进行掺杂,对其导电性和热电性能进行了计算分析比较[8].樊东晓等通过实验,采用甩带法得到快凝高锰硅合金粉末,用Ge对Si位的取代使得衍射峰位向低角区偏移,结果提高了材料的电导性和热电性能[9].王立等人采用基于密度泛函理论的第一性研究方法,对未掺杂以及B掺杂Mn4Si7的电子结构和光学性质进行了研究,结果发现B掺杂引起Mn4Si7的折射率,吸收系数,反射率,反射系数及光电导率都有所增加[10]. Chen等通过固相反应,球磨,火花等离子烧结等方法制备了多晶高锰硅化物,研究Al和(Al, Ge)掺杂对样品微观结构和热电性能的影响发现Al掺杂能够导致电导率和功率因数的提高[11].
目前,虽然已有一些关于Al,Ge掺杂高锰硅化物的研究,但大多都是基于实验上探讨,在理论方面关于Al,Ge掺杂高锰硅化物的研究还比较少,因此,本论文采用基于密度泛函理论(Density functional theory,DFT)的第一性原理计算方法计算分析了Ge,Al分别掺杂,以及共掺杂Mn4Si7的能带结构、态密度和光学性质,从而进一步为研究高锰硅化物应用于光伏材料提供依据.
2 理论模型及计算方法
2.1 建立理论模型
根据无机结构数据库(ICSD)[12],可以看出Mn4Si7晶胞为简单立方烟囱梯状结构,其空间点群为P4-c2,晶格常数a=b=0.5525 nm,c=1.7463 nm,晶面角α=β=γ= 90°.每个晶胞中有4个原胞,Mn4Si7晶胞中一共有44个原子,其中Mn原子16个,占据5个位置,Si原子28个,占据4个位置.本论文采用1x1x1的纯Mn4Si7作为基体,分别用Ge掺杂取代Mn4Si7中的一个Si原子,用Al掺杂取代Mn4Si7中的一个Mn原子,以及用Ge-Al共掺杂取代Mn4Si7中一个Si原子和一个Mn原子.图1(a)为Mn4Si7晶体结构,图1(b-d)分别为Ge,Al,以及Ge-Al共同掺杂Mn4Si7的晶体结构.
2.2 计算方法
本论文计算采用CASTEP模块[13],CASTEP模块是特别为固体材料学而设计的一个现代的量子力学基本程序,其运用密度泛函理论(DFT)平面波赝势方法,进行第一性原理量子力学计算,来探索如半导体,陶瓷,金属,矿物和沸石等材料的晶体结构和其他性质[14].运用基于密度泛函理论(DFT)处理离子与电子之间的相互作用,利用广义梯度近似(generalized gradient approximation,GGA)的PBE(Perdew,Burke and Ernzerhof)[15]基组处理原子间的交换关联能,分别对本征态Mn4Si7及Ge-Al分别掺杂和共掺杂的晶胞进行几何结构优化,得到稳定的几何结构,然后对稳定的几何结构进行结构和性质上的计算分析.设定平面波的截断能(Cut off)为400 eV,k点取样选为4x4x1,在迭代的过程中单原子能量收敛精度为2.0x10-6eV/atom,最大应力0.1 GPa,原子间最大位移的收敛标准为0.002 Å,用于计算的价电子分别为Mn-3d54s2,Si-3s23p2,Ge-3d104s24p2,Al-3s23p1.
3 计算结果分析
3.1 优化后的几何结构
对本征态Mn4Si7,和用Ge,Al单掺杂以及共掺杂的Mn4Si7晶胞进行几何优化,计算结果的晶胞几何参数和总能量如表1所示,根据表中数据可以看出,优化后的晶格参数与实验值相比,平衡晶格常数误差小于1%,体积误差小于1%,说明该计算方法可靠,这个方法可以用于计算其他性质.本征态的Mn4Si7的晶格体积为0.5446 nm3,Ge掺杂后的晶格体积为0.5604 nm3,Al掺杂后的晶格体积为0.5657 nm3,Ge,Al共掺杂后的晶格体积为0.5700 nm3,说明了掺杂原子后晶格晶格常数和体积都有所增大,根据量子化学的观点,这是因为在进行掺杂的过程中,在一定程度上破坏了晶格周期性,使得晶格部分发生了变化,从而晶格常数和体积增大.本征态的Mn4Si7晶体的总能量为-50596.7685 eV,Ge掺杂后的总能量为-52966.1994 eV,Al掺杂后的总能量为-47843.3283 eV,Ge,Al共掺杂后的总能量为-50210.3831 eV,根据能量越低越稳定的化学性质,可以说明掺杂Ge原子的Mn4Si7晶体结构最稳定.

图1 晶体结构 (a)未掺杂;(b)Ge掺杂;(c)Al掺杂;(d)Ge-Al共掺杂Fig. 1 Crystal structures: (a) Undoped; (b) Ge doped; (c) Al doped; (d) Ge-Al co-doped

表1 掺杂前后的晶格常数和总能量
3.2 能带结构和态密度
图2是本征态Mn4Si7和Ge,Al单掺杂以及共掺杂后的能带结构图,本论文主要截取-2 eV~2 eV的能带范围,能量零点为费米能级.为了进一步分析Ge和Al掺杂对Mn4Si7能带结构的影响,首先计算了本征态Mn4Si7的能带结构,如图2(a)所示,从图中可以看出,本征态Mn4Si7的导带底和价带顶都位于G点,能隙为0.810 eV,可知本征态Mn4Si7为直接带隙半导体,这与Allam等人[15]的计算结果基本保持一致.图2(b)为Ge掺杂后的能带结构,可以看出能隙稍微变窄,费米面附近的能带结构发生了变化,导带向低能级方向偏移,价带向高能带方向偏移.图2(c)和(d)分别为Al掺杂和Ge,Al共掺杂的能带结构,从图中可以看出,掺杂后能隙明显减小,说明掺杂后电子从价带跃迁到导带更加容易,导电性能变好,导带微微向低能级方向移动,导带和价带数目明显增多,且在费米能级附近出现了明显的杂质能级,一般称这些杂质能级为类sp带,这是因为当发生原子替换时,在整个体系中会存在大量的自由电子来充当载流子,从而产生了杂化能级,可以大大改善Mn4Si7的光学性能.
图3为本征态Mn4Si7和Ge,Al掺杂的态密度图.图3(a)为本征态Mn4Si7的态密度图,从图中可以看出本征态Mn4Si7的态密度主要是由Mn-3d态和Si-3p态构成,在能量为-6 eV到-2 eV的低能区域时,态密度主要由Si-3p态构成,在能量为-2 eV到2 eV,也即在费米能级附近时,态密度主要由Mn-3d态构成,且费米能级附近有两个明显的尖峰,两个尖峰之间的距离称为赝能隙,赝能隙越宽,表明物质的共价性越强,所以这个赝能隙主要是由过渡金属局域Mn-3d电子轨道提供的,其中导带和价带主要由Mn-3d态和Si-3p态构成,在费米面附近态密度突然下降,表现出明显的半导体性质.从图3(b)可以看出,Ge原子的掺杂对价带的贡献比较大,在能量为-6 eV到-2 eV时,态密度主要由Mn-3d态,Si-3p态和Ge-4p态构成,在能量为-2 eV到2 eV(费米能级附近时),态密度主要由Mn-3d态,Si-3p态和Ge-4s态构成.图3(c)为Al原子掺杂Mn4Si7的态密度图,可以看出Al原子的掺杂对导带的贡献比较大,其中价带主要由Mn-3d态,Si-3p态和Al-3p态构成,导带主要由Mn-3d态,Si-3p态和Al-3s态构成,Al原子掺杂使能隙大大减小了0.792 eV.图3(d)为Ge,Al共掺杂Mn4Si7的态密度图,共掺杂的态密度主要由Mn-3d态,Si-3p态,Ge-4s态和Al-3p态构成,共掺杂比单掺杂产生的杂质能级更多,禁带宽度变窄,掺杂后态密度微微向能量正的方向移动,而且下降坡度变缓慢,说明掺杂后电导率增强,这也与图2 能带结构分析结果保持一致.

图2 Mn4Si7费米面附近的能带结构图:(a)未掺杂;(b)Ge掺杂;(c)Al掺杂;(d)Ge-Al共掺杂Fig.2 Band structures of Mn4Si7 near Fermi plane: (a) Undoped; (b) Ge doped; (c) Al doped; (d) Ge- Al co-doped

图 3 掺杂前后Mn4Si7的态密度:(a)未掺杂;(b)Ge掺杂;(c)Al掺杂;(d)Ge-Al共掺杂Fig. 3 Density of states of Mn4Si7 before and after doping: (a) Undoped ; (b) Ge doped;(c) Al doped ;(d) Ge-Al co- doped
3.3 光学性质
3.3.1复介电函数
复介电函数是研究光学性质的常用指标[16],从介电函数中可以获得固体的能带结构信息和各种其他的光学信息,复介电函数表达式为:
ε(ω)=ε1(ω)+ε2(ω)
(1)
其中,ω为入射光子的频率,ε1(ω)和ε2(ω)分别为复介电函数的实部和虚部,介电函数实部曲线下降的速率决定虚部峰值的高低,虚部对应着光的吸收状况.如图4(a)和(b)分别为Mn4Si7和Ge,Al掺杂Mn4Si7的复介电函数.由图4(a)可知本征态Mn4Si7的介电常数为26.80,Ge掺杂Mn4Si7的介电常数为27.80,Al以及Ge,Al共掺杂后的静电常数明显增大,本征态Mn4Si7在能量为0.748 eV时出现峰值为28.30,Ge掺杂Mn4Si7在能量为1.76 eV时出现峰值14.80,Ge原子和Al原子的介电函数主要由Ge-3d和Al-3p电子结构之间相互杂化形成,电子跃迁能力和概率都增大,掺杂使得介电函数峰值都向高能量方向偏移,导致光产生的电场强度会增大,更有利于提高光伏发电效率.由图4(b)可得,本征态Mn4Si7和掺杂Ge的介电函数虚部基本重合,Al以及Ge,Al共掺杂后的介电函数虚部明显增大,在光子能量为1.67 eV时,出现第一个峰值14.9,掺杂后峰值向左偏移,说明了Al掺杂和Ge,Al共掺杂产生了杂质能级,减小了电子跃迁时所需要的能量,提高了光跃迁强度,光子能量在0到12 eV之间,介电函数虚部都不为零,说明在这个区间内都有电子发生跃迁.
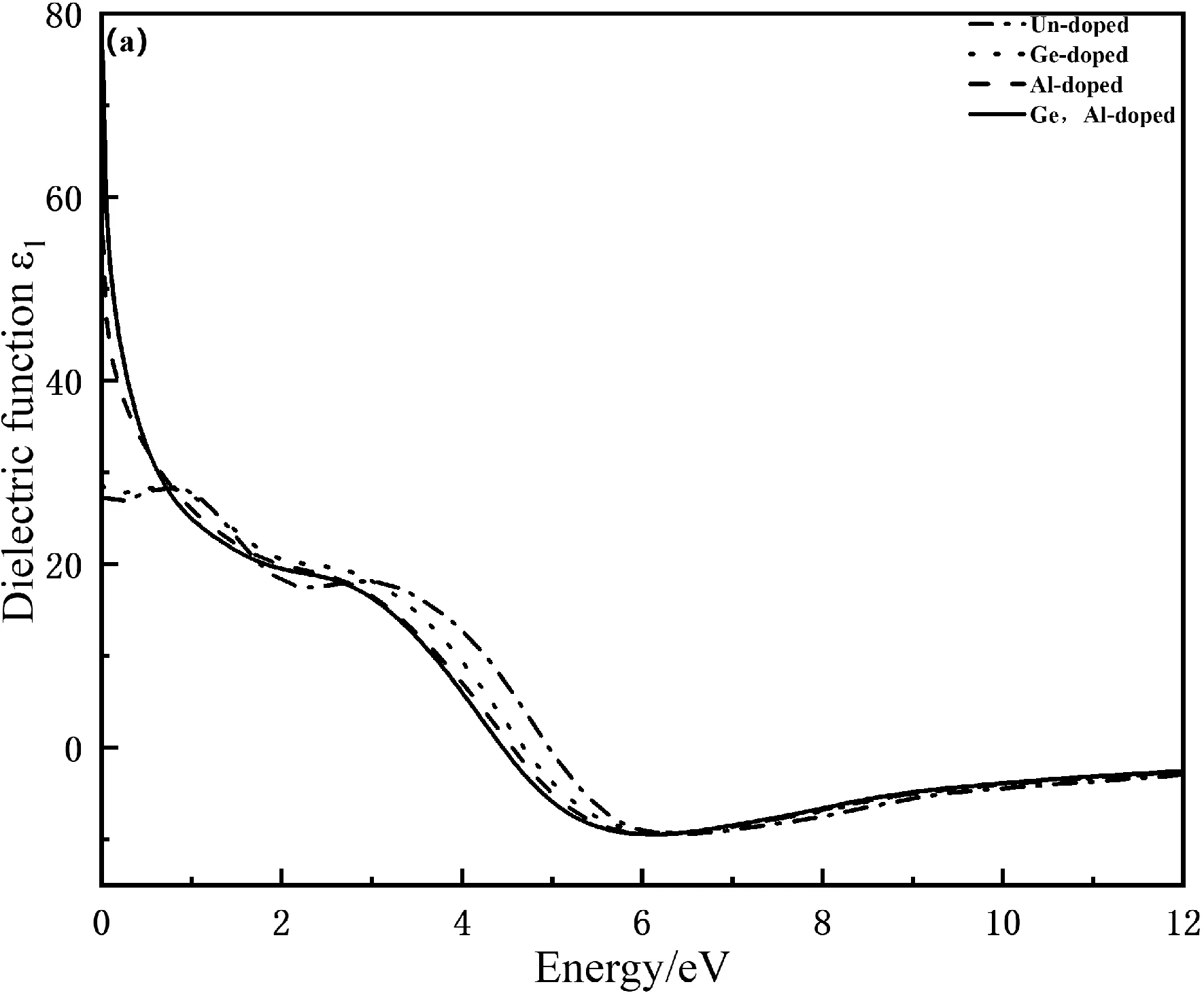
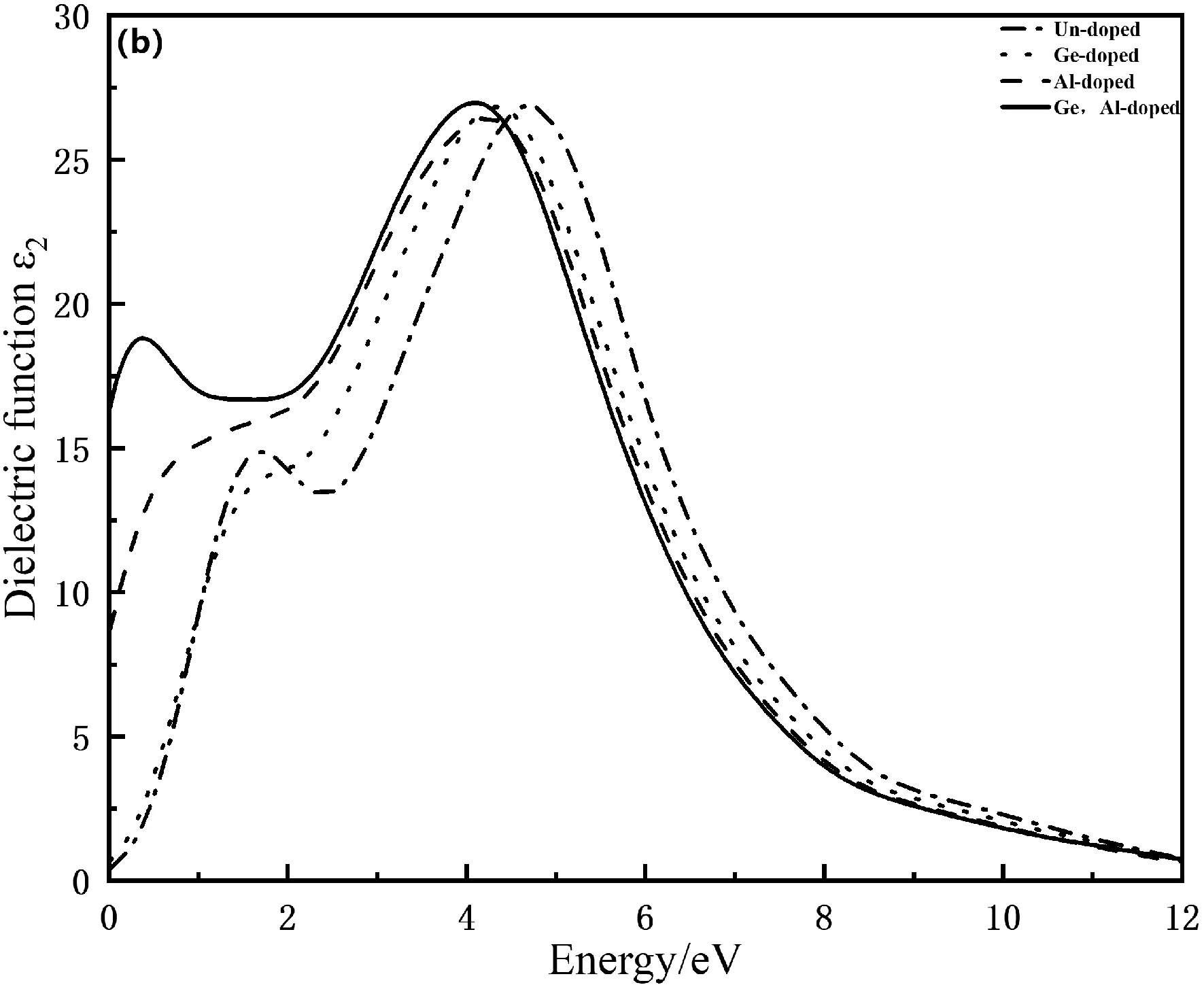
图4 掺杂前后Mn4Si7复介电函数的实部和虚部Fig. 4 The real and imaginary parts of the complex dielectric function of Mn4Si7 before and after doping
3.3.2光电导率
光导电效应是指在光的作用下,体系对电荷的传导率大大提升的现象,光电导率主要与载流子的密度和迁移率有关[17].图5(a)和(b)分别为Mn4Si7和Ge,Al掺杂Mn4Si7的光电导率的实部和虚部,由图5(a)可知,在0到5 eV低能区域,本征态Mn4Si7和掺杂后的Mn4Si7的光电导率都随着光子能量的增大而增大,在光子能量为5.04 eV时,本征态Mn4Si7到达峰值15.80 fs-1,在光子能量为4.71 eV时,Ge掺杂的峰值为14.80 fs-1,在光子能量为4.60 eV时,Al掺杂的峰值为14.30 fs-1,在光子能量为4.47 eV时,Ge,Al共掺杂的峰值为14.20 fs-1,可以看出掺杂后光电导率峰值微微减小且向低能量方向移动.由图5(b)可知,本征态Mn4Si7和掺杂后的Mn4Si7峰值基本重合,可以看出掺杂后光电导率会稍微增大且向低能量方向移动.
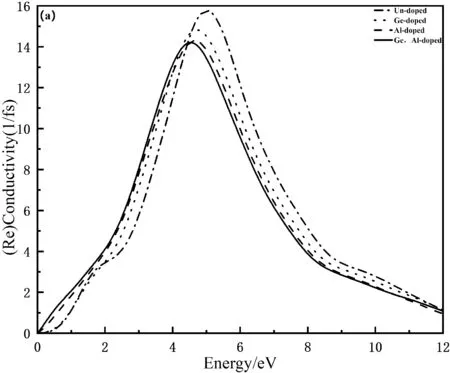

图5 掺杂前后Mn4Si7光电导率的实部和虚部Fig. 5 The real and imaginary parts of the photoconductivity of Mn4Si7 before and after doping
3.3.3反射谱和吸收谱
反射谱是一种反射光谱,是通过光在检验物质表面反射测其反射光线的光谱,而吸收谱则是让光通过物质,然后通过光谱缺失来检验物质性质.如图6(a)和(b)分别为Mn4Si7和Ge,Al掺杂Mn4Si7的反射谱和吸收谱,由图6(a)可以看出,本征态Mn4Si7在光子能量为0时,反射率为0.461 ,Ge掺杂的反射率为0.471,Al掺杂的反射率为0.587,Ge,Al共掺杂的反射率为0.636,说明掺杂后反射率增大.由图6(b)可知,在低能区域,掺杂前后Mn4Si7的吸收系数基本保持一致,掺杂后吸收峰稍微有所减小,说明掺杂后需要电子跃迁的能量减少,吸收系数随着光子能量的增大而增大,当光子能量达到7.18 eV时,达到峰值3.63×105cm-1,之后逐渐减小.

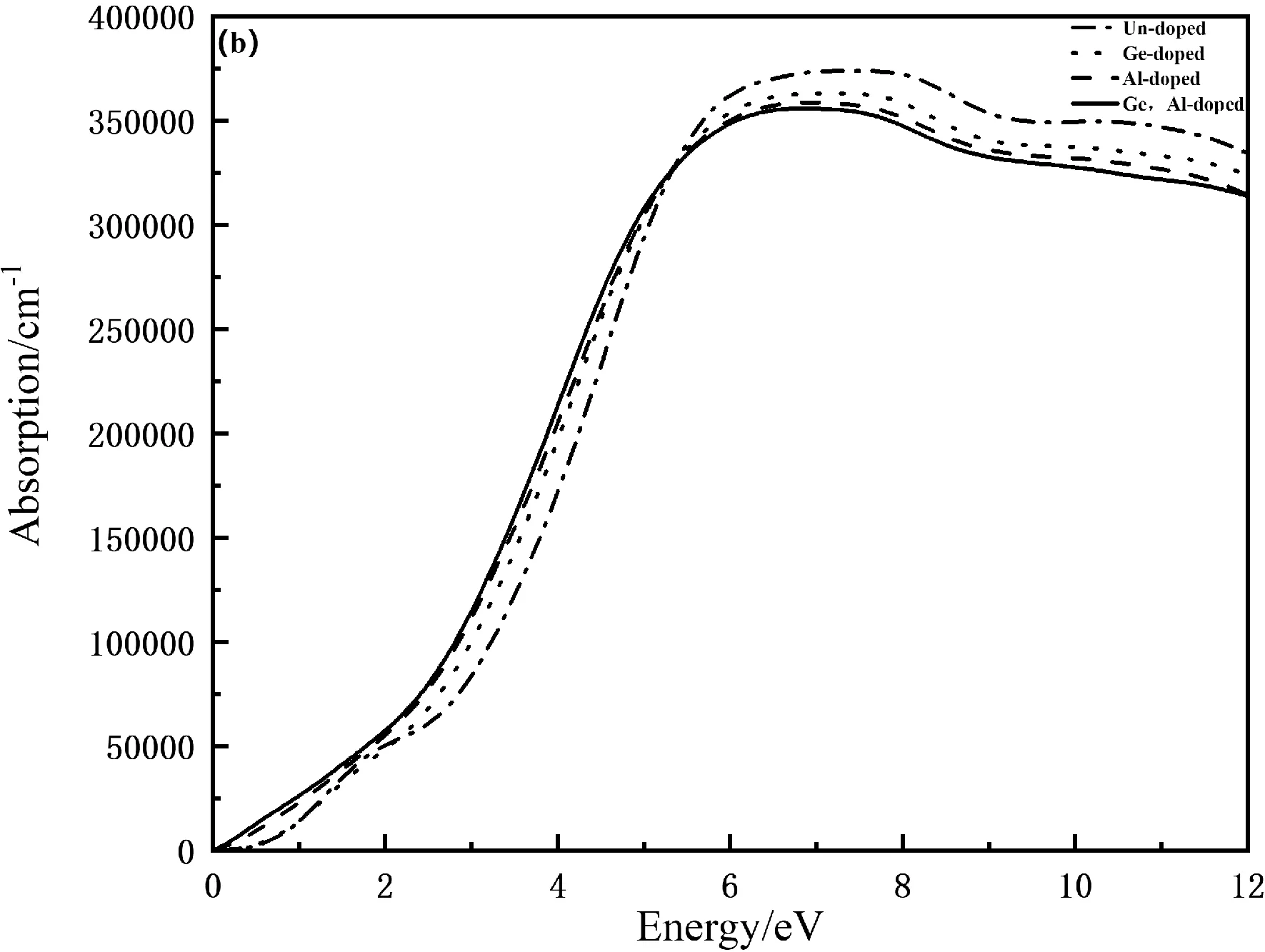
图 6 掺杂前后Mn4Si7的反射和吸收图谱Fig.6 Reflection and absorption spectra of Mn4Si7 before and after doping
3.3.4复折射率
折射率是与反射率相对应的光学参数,根据复介电函数和复折射率之间的关系有:
ε1(ω)=n(ω)2-k(ω)2
(2)
ε2(ω)=2n(ω)k(ω)
(3)
其中,ω为入射光子的频率,n(ω)为复折射率的实部,k(ω)为复折射率的虚部.如图7(a)和(b)分别为Mn4Si7和Ge,Al掺杂Mn4Si7的折射率和消光系数,由图7(a)可知,本征态Mn4Si7的折射率为5.18 ,这与刘等[18]人的计算结果基本一致,Ge掺杂的折射率为5.28,Al掺杂的折射率为7.50,Ge,Al共掺杂的折射率为8.77,可以看出掺杂Ge后稍微有所增加,但Al掺杂,以及Ge,Al共同掺杂后折射率有明显的增大,说明了Ge,Al掺杂可以使得Mn4Si7的折射率增大由图7(b)可知在光子能量为5.64 eV时,本征态Mn4Si7到达峰值3.82,在光子能量为5.27 eV时,Ge掺杂的峰值为3.76,在光子能量为5.23 eV时,Al掺杂的峰值为3.78,在光子能量为5.11 eV时,Ge,Al共掺杂的峰值为3.78,可以看出掺杂后消光系数几乎在同一点达到相同的峰值,与本征态Mn4Si7相比,掺杂后消光系数峰值向低能量方向偏移.

6 结 论
本论文运用基于密度泛函理论的第一性原理计算方法,对比计算分析了本征态Mn4Si7和Ge,Al单掺杂以及共掺杂Mn4Si7的晶体结构,能带结构,态密度和光学性质,得到了以下结论:
(1)从晶体结构可以看出,Ge,Al单掺杂以及共掺杂Mn4Si7的晶格常数和晶胞体积都比本征态的Mn4Si7有所增大.
(2)从能带结构可以看出,本征态Mn4Si7为直接带隙半导体,能隙为0.810 eV,掺杂后体系的能隙减小,特别是Al以及Ge,Al共掺杂时,能隙明显减小,且产生了杂化能级,此时电子从价带跃迁到导带所需的能量最少,电导性最好.
(3)从态密度可以看出,掺杂后由于出现的杂质能级,费米能级附近的态密度不为零,峰值变得更加平缓,电子局域性降低,原子间结合力增强,更加稳定.
(4)从光学性质可以看出,掺杂Ge,Al原子后,在低能区域的很多光学性质产生了不同程度的变化,通过对介电函数的分析发现,掺杂使得介电函数峰值都向高能量方向偏移,导致光产生的电场强度会增大,有利于提高光伏发电效率.通过对光电导率的分析发现,掺杂后光电导率峰值微微减小且向低能量方向移动,更有利于光子的跃迁.通过对反射谱和吸收谱的分析发现,在低能区域,掺杂后反射率增大,吸收峰微微向低能方向移动,吸收系数增大.通过对复折射率的分析发现,掺杂后折射率增大,消光系数峰值向低能量方向偏移.总之,掺杂Ge-Al后,Mn4Si7的光学性质得到了明显的改善.
