人工智能预测药物-靶标相互作用研究进展
2022-02-23李擎宇张孝昌王升启
李擎宇,张孝昌,王升启
(军事科学院军事医学研究院辐射医学研究所,北京 100850)
药物靶标(通常是蛋白质)是指体内与特定疾病过程具有内在联系、可通过与药物作用从而产生预期治疗效果的分子[1]。新药研发的首要问题就是对药物-靶标相互作用(drug-target interaction,DTI)的鉴定,即确定药物分子和靶标之间是否会产生相互作用,并基于此寻找能够作用于特定靶标的药物分子。同时,DTI鉴定也是多重药理和药物重定位等研究的基础。研究发现,复杂疾病往往是由多个基因、多种因素造成的[2],而一种药物也往往具有多种功能、多个靶标[3]。本研究团队早在2000年就提出“中药基因组学”和“中药化学组学”的研究路线[4],并在随后的一系列研究中系统揭示了复方丹参方“多成分-多途径-多靶点”治疗血瘀证的分子机制[5-12]。在此基础上,我们提出“药靶组学”概念[13](图1),为研究药物和疾病之间的关系提供新策略。

图1 药靶组学概念.药靶组学是通过多组学融合等手段鉴定人体可用于药物作用并改善健康的所有效应分子集合.
药物研发过程耗时长、成本高且伴随一定的盲目性。通常新药从研发到上市需要10~15年[14]。据估算,现今一个新药研发成本已达30亿美元[15]。尽管投入大量时间和经费,药物研发的成功率却很低,临床阶段的总成功率仅为10.4%[16]。导致以上结果的原因之一是药物靶标和化学分子数目庞大,而具有相互作用的“药物-靶标对”却很少。据估计,在研发新药时需要考虑的有机分子数量超过1×1060个[17]。截至2015年,美国FDA批准的来源于人或者病原体的靶标共计893个[18]。而一项调查显示,人体内有5000~10 000个潜在的靶标[19]。大量的待研究数据使药物研发伴随着一定的盲目性,也使研发时间和研发成本居高不下。高效鉴定DTI是加速药物研发的关键。
人工智能(artificial intelligence,AI)是计算机学科的分支,是一门用计算机来研究和模拟人类智能的学科,擅长从庞大复杂的数据中挖掘出信息和规律。与传统计算方法相比,AI非常适合筛选大型化学数据库[20]。利用AI辅助DTI预测的研发策略具有速度快、效能高的特点,能够加快研发速度,降低研发成本。常用的模型有支持向量机(support vector machine,SVM)[21-23]、决 策 树(decision tree)[24]、卷积神经网络(convolutional neural network,CNN)[25]、长短期记忆网络(long short-term memory)[26]和生成对抗网络(generative adversarial networks)[27]等。
目前,AI已经被一些企业投入到实际应用中。Atomwise公司长期致力于用AI辅助药物研发,可对大量化合物进行筛选,从而识别和预测以高亲和力结合的药物分子和药物靶标,处于世界领先地位。其开发的深度CNN——AtomNet[28]可以对小分子和靶标蛋白的相互作用进行预测。Atomwise公司通过与斯坦福大学、哈佛大学和制药公司合作,已为27种疾病的潜在药物研发提供了协助[15,29]。Exscientia 公司开发了 Centaur Chemist平台,利用大数据和AI针对特定靶标蛋白设计和筛选小分子化合物,为临床试验提供候选药物分子。2019年,Exscientia与GlaxoSmithKline公司合作,依托Centaur Chemist平台研发了治疗慢性阻塞性肺病的候选药物,大幅度提高药物研发效率。诸多成功案例表明,相对于传统实验手段,AI主导的DTI预测在大体上可靠且有效,具有极大的发展潜力。本文对用于DTI预测的AI方法进行归类和综述,以期为药物研发中的模型开发和实际应用提供参考。
1 药物-靶标相互作用预测方法
在新药研发过程中,DTI的鉴定是一个非常复杂的步骤,它对于候选药物的发现、药物分子作用机制的理解、药物分子的多靶标研究和药物重定位等问题具有重要意义。从海量的靶标蛋白和数据量庞大的化学分子库中挖掘出含有疾病治疗价值的信息是研究者们迫切想要解决的问题。如图2所示,现今国内外DTI预测方法可分为基于配体的方法[30]、基于结构的方法[31-33]和基于化学基因组学的方法[34]。本文综述的AI预测模型可归类为基于化学基因组学的方法,下面首先简述该3类DTI预测方法的基本思想和原理。
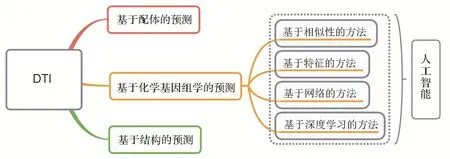
图2 药物-靶标相互作用(DTl)预测方法.
1.1 基于配体的预测方法
基于配体的预测方法(图3)是利用特定靶标的已知配体信息,预测新配体与靶标之间相互作用的方法[35],用于预测分子在特定靶标上的生物活性。最常用的基于配体的预测方法为定量构效关系(quantitative structure-activity relationships,QSAR)方法[36],根据化合物的结构和活性建立模型,以定量的形式来研究分子和靶标的相互作用。具体而言,该方法基于分子活性与分子结构相关的假设,即结构相似的分子通常有相似的活性,通过对比分子与特定靶标的已知配体的相似性来评估分子和靶标的相互作用[37]。QSAR方法可以在靶标结构未知的情况下,对分子进行有效筛选,其缺陷在于回归方程的物理意义模糊,无法帮助理解分子和靶标的作用机制。此外,在已知配体信息不足时,模型的性能会受到限制。
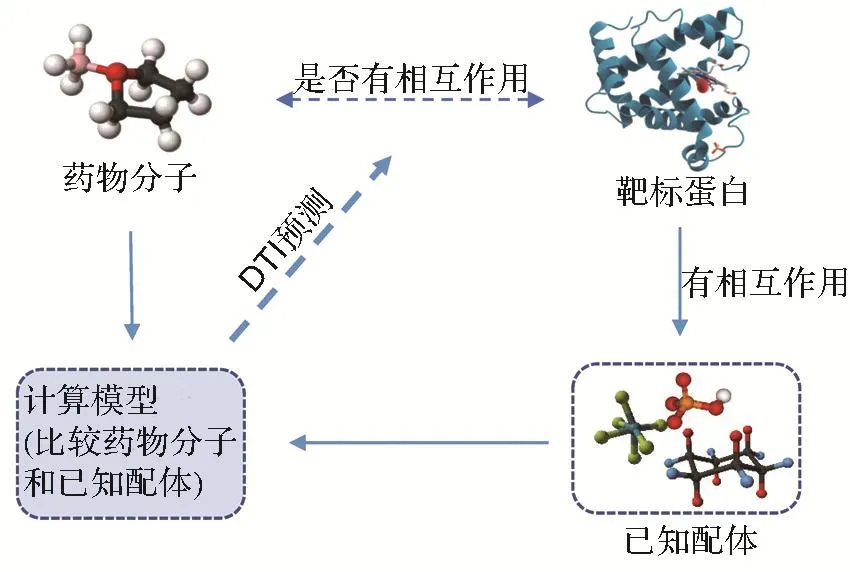
图3 基于配体的DTl预测方法.
1.2 基于结构的预测方法
基于结构的DTI预测方法是根据药物分子和靶标蛋白的空间结构来对二者的相互作用进行预测,其中最著名的方法为分子对接方法。分子对接是基于结构匹配和能量匹配原则,研究受体和药物分子之间的结合模式和亲和力的一种理论模拟方法[38]。分子对接方法有很多种,例如Rarey等[39]提出的DOCK方法,Gohlke等[40]提出的DrugScore方法和Wang等[41]提出的SCORE方法。常见的分子对接方法可以分类为柔性对接、半柔性对接和刚性对接。分子对接方法具有运算速度快、预测准确的特点,但该方法依赖药物分子和靶标蛋白的结构,在结构信息未知的情况下无法使用[42]。例如,对于目前已知结构很少的G蛋白偶联受体,此法则难以应用[21]。
1.3 基于化学基因组学的预测方法
基于化学基因组学的DTI预测方法将化合物空间和靶标蛋白的基因组空间结合起来,构造了药理学空间,充分利用丰富的生物化学多组学数据预测DTI[43]。具体而言,该类方法通过提取药物分子和靶标蛋白的生物学特征或其描述符作为输入,基于复杂的预测模型来预测二者的相互作用。基于化学基因组学的预测方法有3种思路:基于靶标家族的配体学习、基于配体结合位点的靶标共有配体推断和基于配体-受体相互作用的药物-靶标关系预测[21]。该类方法的优势在于有大量的公开生物学数据可供使用,其缺陷在于当有大量的“药物-靶标对”之间的相互作用未被确证时,其预测性能将会受限[43]。基于化学基因组学的预测方法非常适合与AI模型结合,因为药理学空间数据量庞大,而AI可快速、有效地从海量数据中提取信息。
2 人工智能在药物-靶标相互作用预测中的应用
DTI预测的核心问题是判断药物分子和靶标蛋白是否会产生相互作用。AI可以基于已有的药物分子与靶标蛋白相互作用的信息,对未知的药物分子和靶标蛋白进行预测,从而筛选药物分子,继而能够快速、有效地为后续临床试验确定候选药物。总体而言,应用在DTI预测的AI方法可以分为4类:基于相似性的方法、基于特征的方法、基于网络的方法和基于深度学习的方法。
2.1 基于相似性的方法
基于相似性的方法认为,如果药物d与靶标t可以相互作用,那么与药物d相似的分子可能会与靶标t产生相互作用;与t相似的靶标可能会与药物d相互作用。基于这种假设,该方法对靶标对或者药物分子对进行相似性评分,再根据评分结果来预测DTI。这类方法往往会根据模型需要来定义一种度量,用于描述相似性。药物分子对的相似性度量可以分为5种类型:基于化学信息、基于配体信息、基于表达信息、基于副作用和基于注释,不同类型的度量有不同的侧重点[44]。Shi等[45]基于相似性排序开发了一个非参数、惰性的预测方法SRP(similarityrank-based predictor),利用相似性和相似性的排序来计算2个指数——趋势指数和反向趋势指数,分别表示药物和靶标产生相互作用的概率和不产生相互作用的概率,在基准数据集上取得了较高的准确率。使用单一的相似性会使模型具有局限性,集成多种相似性度量则会缓解局限性。Perlman等[44]提出了一个结合多种相似性度量来预测药物靶标的框架SITAR(similarity-based inference of drug-targets),此框架中包含了一个新的药物-基因关联程度评价方案。作者对现有的几百种药物应用SITAR进行测试,结果表明该模型明显优于其他模型。在庞大的药理学空间中,只有少部分分子和靶标的相互作用被鉴定出来,如果只用这些数据来训练模型,会让模型的泛化能力降低。为了缓解这个问题,Xia等[46]使用一种半监督学习方法——Laplacian正则化最小二乘法,基于化学结构和基因组序列的相似性来预测DTI。
基于相似性的方法是应用最多的一类预测方法,其优势在于:①不必进行复杂的特征提取和特征选择;②计算化学结构相似性以及基因组序列相似性的度量的发展较为完善;③相似性方法可以直接与核方法相结合;④相似性度量揭示了药物和基因的联系[47]。该类方法的缺陷在于模型的结果和表现会依赖于相似性度量的选取,这是因为相似性定义了药物在高维空间中的相对距离,使用集成学习方法,融合多种度量是解决此缺陷的主要思路。
2.2 基于特征的方法
基于特征的方法是将药物分子和靶标蛋白的生物学信息进行编码,生成特征描述符(descriptors)用以描述分子和靶标,并将这种描述符整合成特征向量,然后应用AI算法进行预测。如图4所示,常用的有SVM、决策树和随机森林等机器学习算法。例如,Cao等[48]应用随机森林方法,结合化学、生物和网络特征的计算方法预测DTI,并且应用在人体内含有的4类DTI网络(酶、离子通道、G蛋白偶联受体和核受体)上,用以验证模型性能。在基于特征的预测方法中最常用的是SVM模型,研究者们一般会通过改进核函数来提高SVM的性能。Jacob等[21]针对受体来构造核函数,应用SVM模型来预测配体-蛋白质相互作用,结果显示模型具有较好的效果,即便是对未知3D结构或者已知配体较少的靶标也有很好的表现。Faulon等[49]结合了多种核函数的思想,构建了一种新的核函数,并以此建立了SVM模型,结合蛋白质序列信息和化学信息来预测DTI。除SVM模型外,一些核方法也被研究者们用来预测DTI。Van Laarhoven等[50]根据已知的药物和靶标相互作用的信息,构建了药物靶标相互作用网络,定义了GIP核(gaussian interaction profile kernel),并采用正则化最小二乘法来预测DTI。

图4 基于特征的DTl预测方法.
基于特征的模型是在机器学习领域中被广泛应用的预测模型,它可以根据药物和靶标特征直接预测DTI,具有很好的预测效果和较好的可解释性。该类方法的缺陷在于为了提高模型性能,在训练之前需要进行特征提取和特征选择,过程往往非常复杂,需要消耗大量的时间。
2.3 基于网络的方法
在生物学研究中,经常使用网络图来表示复杂的生物学过程,如KEGG数据库通过网络图来表示和研究人体内的代谢通路[51]。STRING数据库依据直接相互作用和共表达相关性等构建蛋白质相互作用网络[52]。基于网络预测DTI的方法(图5)也是应用了类似的研究思路,利用相互作用网络挖掘药物分子和靶标蛋白之间的联系,并以此预测DTI。例如,Olayan等[53]开发了一种名为DDR的模型,使用非线性融合方法整合了多种相似性指标,构建了药物靶标的异质网络,并使用随机森林方法从异质网络中寻找药物靶标相互作用对。Thafar等[54]在DDR模型的基础上采用新的特征选择和融合方式,使用图嵌入、图挖掘和机器学习方法预测药物靶标相互作用,构建了DTiGEMS模型。拓扑结构是网络的重要特征,彭利红等[55]综合考虑靶标和药物数据的局部和全局特征,结合靶标蛋白的序列相似性以及药物-靶标网络的拓扑结构性,提出了一种基于局部全局一致性学习的了预测模型。Cheng等[56]提出一种仅使用药物-靶标二分网络拓扑相似性来推断药物分子作用的靶标的方法。但当分子无任何已知的可与其产生相互作用的靶标蛋白时,基于网络的预测方法将会受限。Chen等[57]基于网络的相关性,提出了一种名为NetCBP的半监督学习方法,利用有标签和无标签的数据进行训练,可以缓解数据不足造成的模型性能下降问题。基于网络的方法依赖于训练集数据的质量,数据的噪声和不完整性会对预测结果产生影响。因此,Luo等[58]开发了DTINet方法,不仅整合了一个集成了多种药物相关信息(药物、蛋白质、疾病和不良反应)的异构网络,还通过学习药物和蛋白质特征的低维向量表示来更好地应对有噪声的、不完整的和高维的生物数据。随机游走的方法也可应用于DTI预测,Chen等[59]通过已知的DTI信息,将蛋白质-蛋白质相似性网络、药物-药物相似性网络和药物-靶标相似性网络整合,构建了一个异质性网络,使用随机游走算法提取特征,实现DTI预测。基于网络的预测方法不依赖于分子结构,但是如果在网络中无已知的可达的路径,则无法对“药物-靶标对”进行预测;同时,使用网络方法预测的结果会具有偏差,模型预测结果偏向于已知配体较多的靶标,或已知靶标较多的配体[60]。

图5 基于网络的DTl预测方法.
2.4 基于深度学习的方法
深度学习是一种基于人工神经网络的机器学习算法,在语音识别、图像识别和自然语言处理等领域有着广泛的应用。近年来,应用深度学习方法进行药物研发呈上升态势[61]。基于深度学习的预测方法首先根据药物分子或靶标蛋白的生物学信息来构建特征向量,再将特征向量输入深度神经网络进行训练。Wang等[26]基于位置特异性评分矩阵和勒让德矩提取蛋白质特征,并且与药物分子的结构信息相结合,构建“药物-靶标对”的特征向量,通过构建深度长短时记忆模型对DTI进行预测。Wen等[62]开发了一个深度学习框架DeepDTI,该框架首先利用无监督预训练方法从原始数据中抽取特征表示,然后依照已知的DTI信息来建立分类模型。深度学习方法也可以结合特征提取等方法来对数据降维,加快模型训练。Peng等[63]提出了一种基于表示学习和深度神经网络的方法,该方法首先利用Jaccard相似系数和重启随机游走模型从异构网络中提取药物和蛋白质的相关特征,然后利用去噪自编码器来降维,并构造CNN来预测DTI。Rayhan等[64]使用自编码器和CNN构建FRnet-DTI的编码模块和分类器模块,用于完成预测任务。图神经网络也被广泛应用于药物靶标相互作用预测中。Zhao等[65]使用图卷积网络对相互作用对进行特征提取,随后使用深度神经网络进行预测。深度学习的优势在于不用直接对特征进行筛选或降维,而可以把所有已知的信息作为输入,避免特征信息的损失[66]。
3 人工智能预测药物-靶标相互作用面临的问题和挑战
3.1 模型评价问题
与其他二分类任务相同,DTI预测使用P-R曲线下面积(area under the precision-recall curve,AUPR)和ROC曲线下面积(area under the receiver operating characteristic curve,AUROC)指标来衡量模型性能。为了绘制该2种曲线,需要基于真阳性(true positive,TP)、假阳性(false positive,FP)、真阴性(true negative,TN)和假阴性(false negative,FN)计算假阳性率(FPR)、召回率(TPR)和准确度(precision):
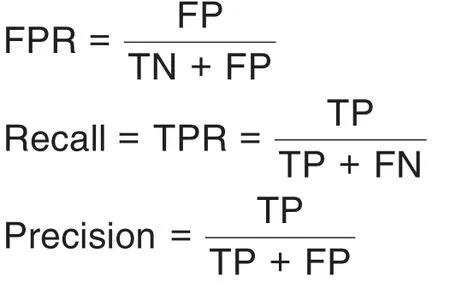
在绘制P-R和ROC曲线之后,计算其下方面积即可分别得到AUPR和AUROC 2个指标,两者数值越大说明模型效果越好。除了AUPR和AUROC 之外,误差率(error rate,ER)也常被用于对比模型性能,其定义为:ER=1-AUPR。
Yamanishi数据集[42]是 DTI预测任务的金标集,按照靶标类型分为4个小数据集,分别是酶、离子通道、G蛋白偶联受体和核受体。本文选取了FRnet-DTI[64],GCN-DTI[65],DDR[53]和 DTiGEMS+[54]4个高性能模型在Yamanishi数据集上的表现进行了对比,对比结果如表1所示。2类方法在不同数据集上均有着优异的表现。基于深度学习的模型在酶数据集上的表现更为优秀,而在其他3个数据集上,基于网络的模型表现更好。在考虑实际应用时,可以根据具体情境的不同,使用多种方法进行预测,以期得到更为可靠的结果。

表1 基于网络和深度学习模型的比较
3.2 负样本问题
负样本问题旨在研究如何界定和构建负样本集用于训练和预测,是DTI预测任务中的重要问题。对于任意一对药物和蛋白质,如果经过实验验证,证实二者存在相互作用,那么二者构成正样本。但是对于未经实验验证的“药物-蛋白质对”来说,难以界定它是正样本还是负样本。负样本集的选择会影响到数据的标签,同时也会决定模型的决策边界,对预测的准确性有很大的影响。
为处理负样本问题,大多数模型采取的策略为:将未经过实验验证的“药物-靶标对”均视为负样本,即将正样本以外的数据均视为负样本,这种做法会导致负样本集中存在假阴性,即混杂了一些正样本,这会导致模型对正样本的鉴定能力降低。Zheng等[67]基于“药物-靶标对”的相似性原则,通过已知的负样本来推测潜在的负样本,构建可靠的负样本集。Wang等[68]使用不同指标衡量蛋白质之间的差异,设计概率模型来构建负样本集。具体方法为:对给定药物和蛋白质,计算其与正样本的差异,由此计算其作为负样本的概率。
负样本问题来源于数据本身,随着时间的推移,越来越多的DTI对被鉴定,该问题即会得到逐步缓解,然而目前只能从数据集构建上寻找策略,这在一定程度上可实现对决策边界的优化,提高预测的准确性。
4 结语
DTI鉴定是药物研发的关键步骤,数量庞大的药物分子和靶标蛋白中蕴藏着丰富的生物学信息,而AI擅长从数据中挖掘知识,因此将二者有机结合,以AI预测DTI,将会提高药物研发效率。目前,诸多的成功案例已经表明,AI方法可以有效的辅助药物研发。用于DTI预测的AI模型可以分为基于相似性、基于特征、基于网络和基于深度学习的方法,这些方法从不同角度寻求替代传统实验的途径,加速药物研发。
总体而言,AI方法在DTI预测中有极大的潜力,但也面临着一些问题和挑战。AI模型通常是在过于简化和理想的条件下构建的,往往不能准确反映实际应用中的问题[69]。在庞大的药理学空间中,只有极少一部分DTI被确证,如何有效的利用已确证和未确证的“药物-靶标对”来训练模型是一个难题。AI不具备良好的可解释性,无法帮助理解药物分子作用机制,导致其可靠性受到质疑。为应对这些挑战,未来AI模型可能更倾向于用半监督学习方式,充分利用各个组学的数据,并结合注意力机制等可解释模块来预测DTI,在保证预测精度的同时辅助相互作用机制的研究。虽然目前的方法还存在一些缺陷,但随着数据的进一步积累和计算能力的进一步增强,在多组学数据融合的药靶研究策略下,AI有望在药物研发领域发挥更大的作用,成为药物研发的“新动力”。
