北京棒杆菌单体天冬氨酸激酶T379N/A380C/T65I/D173G突变体酶学性质表征及工程菌构建
2022-02-16王亚南刘晓婷樊占青王哲人闵伟红
王亚南,刘晓婷,樊占青,王哲人,高 欣,闵伟红
(吉林农业大学食品科学与工程学院,小麦和玉米深加工国家工程实验室,吉林 长春 130118)
天冬氨酸族氨基酸被广泛应用于食品、饲料、化妆品、药物等[1-2],目前主要采用低成本、对环境友好的微生物发酵法生产[3-4]。其代谢途径受天冬氨酸激酶(aspartate kinase,AK)、高丝氨酸脱氢酶[5]、高丝氨酸酰基转移酶[6]和高丝氨酸巯基酶[7]调节,其中AK是影响碳流走向的首个关键限速调控点,将ATP的磷酸基团转移到天冬氨酸(Asp),并生成天冬氨酰-4-磷酸[8],最终转化为赖氨酸(Lys)、苏氨酸(Thr)和甲硫氨酸(Met),因此AK对天冬氨酸家族氨基酸产物的积累具有至关重要的影响。为了优化代谢途径过量积累天冬氨酸族氨基酸,解除变构抑制和提高酶的催化活性成为研究关注点[9-12]。
在已报道的生物体中,AK的晶体结构主要为同型和异型寡聚体[13],存在形式主要有单功能AK(AKI、AKII和AKIII)和双功能AK-HSDH(I和II)[14]。如在拟南芥(Arabidopsis thaliana)中的AK是一种高寡聚体,AKI受Lys和Met的抑制,AKII和AKIII只受Lys抑制[15-17];在大肠杆菌(Escherichia coli)中的AKIII是同源二聚体,受Lys抑制[18];而在谷氨酸棒杆菌(Corynebacterium glutamicum)中的AKII受Lys和Thr协同抑制[19],是由α和β亚基组成的异源四聚体(α2β2),α亚基在N末端包含一个催化结构域,在C末端包含一个调节结构域[20]。以上报道的AK均以多聚体形式存在,然而本实验室发现北京棒杆菌中AK(C. pekinenseaspartate kinase,CpAK)呈单体状态[21],与谷氨酸棒杆菌中AK(C. glutamicumaspartate kinase,CgAK)具有98.78%的同源性,可基于CgAK的序列构建CpAK的模型。
与CgAK相同,CpAK含有调节结构域和催化结构域,包含Thr、Lys、ATP、底物Asp 4个关键配体。目前,多数研究对调节结构域的抑制剂Lys结合位点进行定点突变[22-25],对催化结构域ATP、底物Asp结合位点的改造较少。本研究在各个配体周围对4个关键残基位点同时进行定点突变,筛选出有效解除反馈抑制的酶活力提高突变体T379N/A380C/T65I/D173G,通过无缝克隆技术构建北京棒杆菌工程菌,旨在为构建高产天冬氨酸族氨基酸菌株提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 质粒、菌株与试剂
大肠杆菌重组质粒(pET-28a-AK)、大肠杆菌感受态DH5α和BL21(DE3)、北京棒杆菌AS1.299均由实验室提供;表达载体pEC-XK99E 湖南丰晖生物科技有限公司。
质粒抽提试剂盒、胶回收试剂盒、Taq酶、核酸电泳Marker、DpnI消化酶 TaRaKa(大连)有限公司;丙基硫代-β-D-半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)、卡那霉素、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)试剂盒 北京鼎国昌盛生物技术有限公司;非变性镍柱柱料 美国GE公司;限制性核酸内切酶EcoR I、BamH I、引物合成、测序由生工生物工程(上海)股份有限公司提供;无缝克隆试剂盒MonCloneTMHi-Fusion Cloning Mix V2莫纳(苏州)生物科技有限公司。
1.1.2 培养基
LB液体培养基:1%蛋白胨,0.5%酵母浸粉,1% NaCl;LB固体培养基另加2%琼脂。
北京棒杆菌感受态培养基:在LB液体培养基上另加0.3%甘氨酸,0.1%吐温80。
北京棒杆菌电转化恢复培养基:0.25%酵母浸提物,0.5%蛋白胨,0.5% NaCl,9.1%D-山梨醇,18.5%脑心浸液,固体培养基另加入2%琼脂。
种子培养基:2.5%葡萄糖,2%玉米浆,0.1% KH2PO4,0.5% MgSO4,0.125%尿素。
发酵培养基:10%葡萄糖,2%玉米浆,2% (NH4)2SO4,0.1% KH2PO4,0.05% MgSO4,0.001% MnSO4,0.001% FeSO4,0.000 1% VB1,0.000 6% VB6,0.02% VB12,0.000 01%生物素。
以上培养基均用2 mol/L NaOH溶液调pH值为7.0。
1.2 仪器与设备
梯度聚合酶链式反应(polymerase chain reaction,PCR)扩增仪 德国Eppendorf AG公司;DYCP-31DN核酸电泳仪 北京市六一仪器厂;Spectra Max 190酶标仪美国Molecular Devices公司;超声破碎仪 宁波新芝生物科技有限公司;Z36HK低温高速离心机 德国Hermle公司;蛋白印记转膜仪 美国Bio-Rad公司;SE260蛋白电泳仪、琼脂糖凝胶成像仪 美国GE公司;L-8900高速氨基酸分析仪 日本日立公司。
1.3 方法
1.3.1 突变株的构建
PCR扩增AK基因:运用Primer 5.0软件设计引物,上游引物为5’-GGTCGTGGTGGTTCTNNNACCACTGCAG TTG-3’,下游引物为5’-CGCAACTGCAGTGGTNNNAG AACCACCACG-3’,NNN表示饱和突变,合成于生工生物工程(上海)股份有限公司。从重组大肠杆菌中提取pET-28a-AK质粒并以其为模板,在引物作用下,经94 ℃预变性10 min;94 ℃变性1 min,54~58 ℃退火1 min,72 ℃延伸10 min(以上3 步循环18 次);72 ℃保温10 min完成突变PCR,并进行1%琼脂糖核酸电泳验证。将验证成功的PCR产物用DpnI酶37 ℃金属浴消化2 h。转入大肠杆菌宿主:将2 μL消化后的PCR产物转入冰上预冷30 min的BL21感受态细胞,冰浴5 min,42 ℃热激90 s,再冰上孵育2 min,加入不含卡那霉素的LB培养基900 μL,于37 ℃、170 r/min培养1.5 h,9 000 r/min离心2 min弃上清液800 μL,吹悬菌体后涂布于LB固体平板(含终质量浓度为50 mg/L卡那霉素)上,37 ℃过夜培养。
高通量筛选:将单克隆菌株从抗性平板转接到96 孔板中过夜培养,加入终浓度为1 mmol/L IPTG,在28 ℃、130 r/min培养12 h诱导蛋白表达。3 500 r/min离心45 min后弃上清液,加入100 μL磷酸盐缓冲液反复吹打菌体,冻融破碎后测定其酶活性,高通量筛选出高酶活力的突变株,方法参考文献[26]。以高酶活力菌株扩大培养的菌液为模板,在克隆引物作用下进行菌液PCR扩增,反应条件:94 ℃预变性10 min;94 ℃变性30 s,58 ℃退火60 s,72 ℃延伸90 s,循环30 次;72 ℃延伸10 min。用琼脂糖凝胶电泳进行菌液PCR产物验证,并进行基因测序以证明突变株构建成功。
1.3.2 蛋白表达与纯化
以2%接种量将野生型(wild type,WT)和突变体菌株在100 mL LB液体培养基中培养,当OD600nm达到0.7时,添加IPTG(终浓度1 mmol/L)100 μL,分别在25 ℃和28 ℃培养10 h诱导蛋白表达。4 ℃、8 000 r/min离心10 min,弃上清液并加入预冷的磷酸盐缓冲液重悬菌体,菌体经超声破碎并离心后,上清液经0.22 μm滤膜处理后即为AK粗酶液,将其通过镍柱分离纯化,经不同浓度咪唑溶液梯度洗脱去除杂蛋白后得到AK纯化液。所得样液变性后用SDS-PAGE和Western Blot验证,具体方法参考文献[12]。
1.3.3 酶动力学测定
用10 个不同浓度(0.5、1、3、5、7、9、10、12、14、16 mmol/L)的L-Asp底物进行酶活力测定,并在520 nm波长处测吸光度,反应体系及条件参考文献[24],计算比活力。通过Origin 8.5软件与Hill方程V=VmaxSn/(Kn+Sn)进行非线性拟合。反应速率V表示1 min内1 mg酶的催化能力,Vmax表示酶完全被底物饱和时的最大反应速率。
1.3.4 AK酶学性质分析
最适温度:底物浓度为3 mmol/L,反应体系与酶活力测定相同,在不同温度(15、20、25、26、28、30、35、40、45、50 ℃)条件下反应30 min,最高酶活力定义为100%。
最适pH值:反应体系同上,加入不同pH值的Tris-HCl缓冲液(6.0、6.5、7.0、7.5、8.0、8.5、9.0、9.5、10.0),最高酶活力定义为100%。
稳定性:在得出的最适温度和最适pH值条件下,其余反应体系不变,反应30 min后测定酶活力,0 h酶活力定义为100%,之后每隔1 h测定酶活力,共测10 次。
抑制剂对酶活力的影响:向反应体系中加入不同的抑制剂(Thr、Lys、Met、Lys+Thr、Lys+Met、Thr+Met或Thr+Lys+Met),每种或多种抑制剂的最终浓度分别为0.2、1、5、10 mmol/L,以检测抑制剂对AK活力的影响,添加水的对照组相对酶活力定义为100%。
1.3.5 工程菌构建
获得目的基因片段:提取WT和突变株T379N/A380C/T65I/D173G的质粒为模板,在克隆引物作用下经94 ℃预变性10 min;94 ℃变性1 min,60 ℃退火1 min,72 ℃延伸10 min(以上3 步循环18 次);72 ℃保温10 min完成PCR扩增,并进行1%琼脂糖核酸电泳,再进行胶回收获得大量的目的基因。
线性化载体pEC-XK99E:提取质粒获得表达载体pEC-XK99E,线性化载体的反应体系为EcoR I酶2 μL、BamH I酶2 μL、pEC-XK99E质粒25 μL,将线性化载体进行1%琼脂糖核酸电泳和胶回收。
无缝克隆连接:将胶回收的目的基因片段和线性化载体按以下体系进行无缝克隆连接:线性化载体1 μL、目的基因片段2 μL、Hi-Fusion Cloning Mix V2 2.5 μL,连接成功后转入大肠杆菌DH5α感受态,操作方法同于转入大肠杆菌BL21感受态。
电转化入北京棒杆菌:将提取测序成功菌株的质粒5 μL加入北京棒杆菌感受态细胞,在冰上预冷10 min后全部转入电转杯,用20 kV电击5 ms,加入LBHIS培养基洗涤后,30 ℃、100 r/min复苏2 h,若菌浓度过低可延长复苏时间。将复苏的菌体于8 000 r/min离心2 min后弃去800 μL培养基上清液,剩余200 μL重悬后涂布于北京棒杆菌电转化恢复固体培养基上(含卡那霉素),30 ℃倒置培养36 h,挑菌测序。
1.3.6 发酵及氨基酸含量测定
对WT和构建的T379N/A380C/T65I/D173G菌株进行48 h菌株发酵培养,每6 h取样,取氨基酸含量较稳定的第30小时发酵液进行氨基酸含量测定[27-29]。取2 g样品于20 mL水解管中,加入16 mL 6 mol/L盐酸溶液,真空脱气30 min,充氮封管,110 ℃水解22~24 h,取出冷却开管,用去离子水无损转移到50 mL容量瓶并定容。准确取1 mL水解液于小瓶中,于真空中脱酸抽干,加1 mL水再抽干,再加1 mL水再抽干备用。上机前准确加入1 mL 0.02 mol/L盐酸溶液,充分溶解,用0.22 μm的水相膜滤膜过滤后用高速氨基酸分析仪进行分析。
1.4 数据分析
2 结果与分析
2.1 关键突变位点的确定
本实验室利用SWISS-MODEL构建单体AK的三维结构,使用ATP Bind server和COACH-D预测配体的结合位点,采用Auto Dock对配体进行对接,成功构建的单体AK模型见图1A[21]。利用Clustal X软件对多个不同生物体内的AK进行同源序列比对,用ESPript进行修饰比对结果,红色部分表示高同源性保守位点(图1B)。运用Discovery Studio 4.0软件对AK模型进行分析,选取了不同配体周围的4个位点进行定点突变:抑制剂Lys的结合位点Thr379和Aln380,Thr379与抑制剂Lys的羧基由氢键紧密连接;Ala380位点与Thr379紧密相连,且与抑制剂Lys通过水分子连接(图1C)。Thr65通过氢键与底物Asp相连是关键的结合位点(图1D)。文献报道,甲烷球菌AK(Methanococcus jannaschiiAK,MjAK)(PDB:2hmf)的Asp211[29]和大肠杆菌(PDB:2j0w)的Asp202位点为催化活性中心位点[18],利用SPDB软件对几种模型进行空间结构比对,发现都对应CpAK中Asp173位点,其位于底物Asp和ATP中间(图1E)。
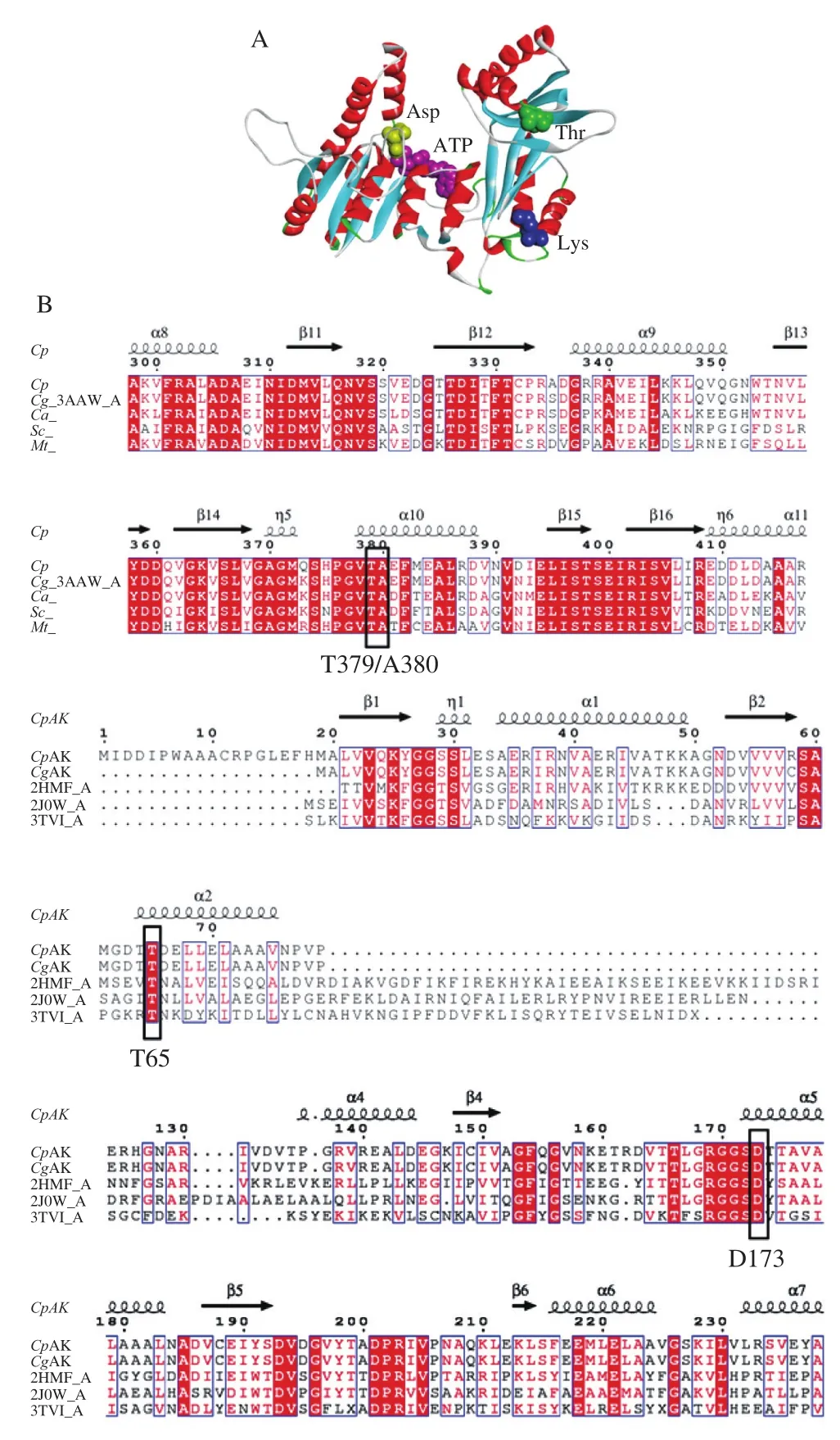
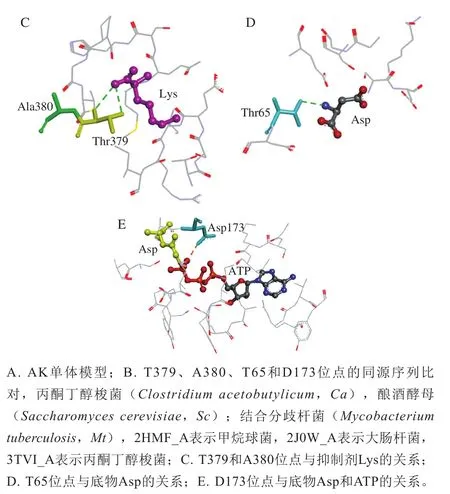
图1 突变位点的确定Fig. 1 Determination of mutation sites
2.2 突变体构建
定点突变PCR扩增产物用1%琼脂糖核酸电泳进行验证,结果如图2A所示,在7 000 bp左右有单一明亮条带,证明目的基因扩增成功。PCR产物经DpnI酶消化作用后用热激法转入到大肠杆菌感受态BL21细胞中,采用高通量筛选方法筛选出高酶活力突变体,并进行菌液PCR,验证结果如图2B所示,在1 000~2 000 bp之间有明显条带,与CpAK(1 317 bp)长度一致,证明目的基因AK已成功导入大肠杆菌菌株。对该菌株进行测序,测序结果与AK原序列比对见图2C,Thr379、Ala380、Thr65、Asp173位点分别由Thr突变为天冬酰胺(Asn)、由Ala突变为半胱氨酸(Cys)、由Thr突变为异亮氨酸(Ile)、由Asp突变为甘氨酸(Gly),成功构建了T379N/A380C/T65I/D173G突变体。
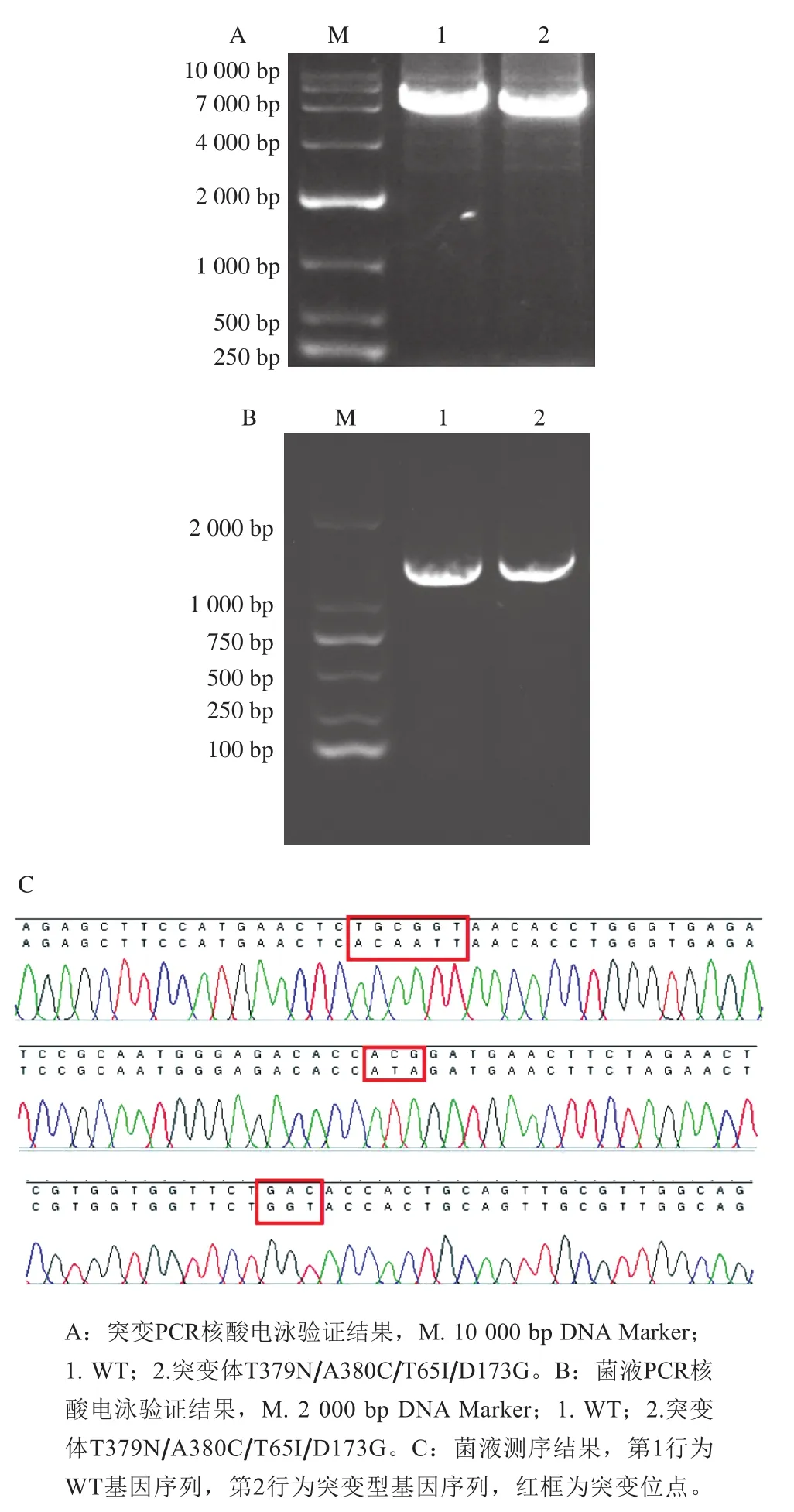
图2 突变体T379N/A380C/T65I/D173G的构建Fig. 2 Construction of mutant T379N/A380C/T65I/D173G
2.3 蛋白表达与纯化
将诱导表达的WT和突变体菌株经破碎、离心、过膜后进行非变性镍柱纯化,得到AK粗酶液和AK纯化液,分别变性后经SDS-PAGE和Western Blot验证,结果见图3,AK纯化液在48 kDa左右处均有单一条带,表明AK蛋白成功表达。
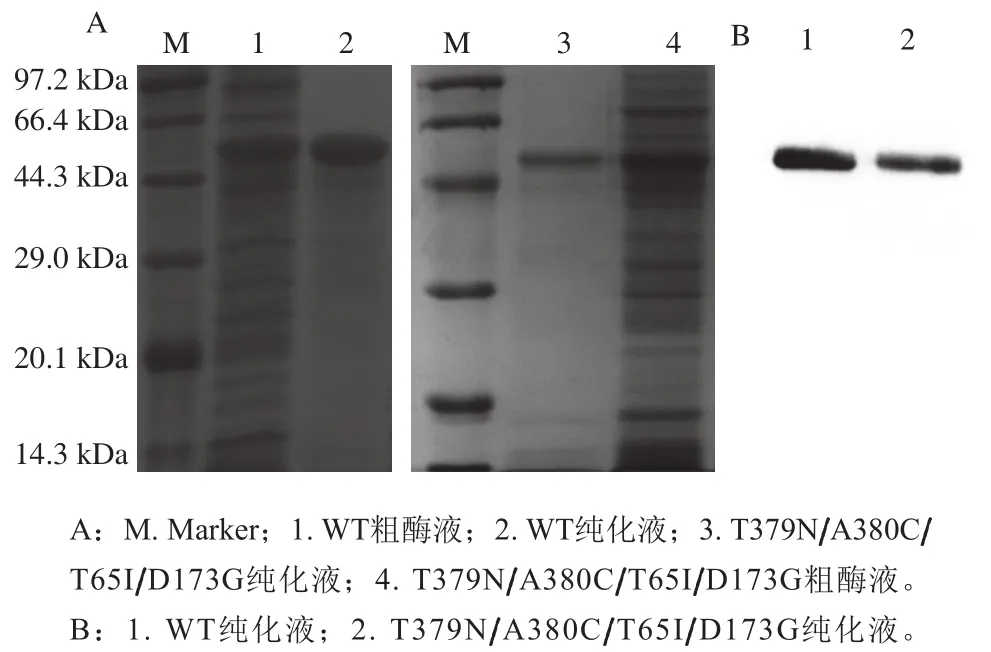
图3 SDS-PAGE(A)及Western Blot(B)结果Fig. 3 Results of SDS-PAGE (A) and Western Blot (B)
2.4 反应动力学分析
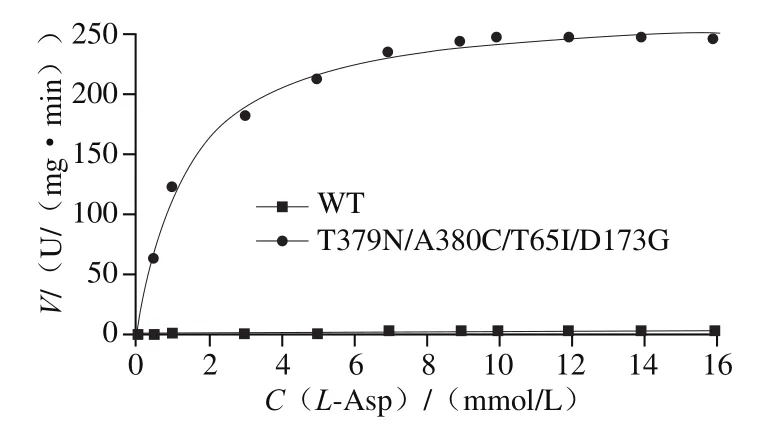
图4 WT和T379N/A380C/T65I/D173G的动力学分析Fig. 4 Kinetic analysis of WT and T379N/A380C/T65I/D173G

表1 WT及T379N/A380C/T65I/D173G动力学参数Table 1 Kinetic parameters of WT and T379N/A380C/T65I/D173G
通过软件Origin 8.5中的Hill方程V=VmaxSn/(Kn+Sn)进行非线性拟合得到Vmax、n和Km,WT和T379N/A380C/T65I/D173G的动力学参数分析如图4和表1所示。T379N/A380C/T65I/D173G的Vmax为267.67 U/(mg·min),WT的Vmax为3.53 U/(mg·min),提高到75.83 倍。Km值表示当反应速率达到最大反应速率一半时的底物浓度,突变后AK的Km值由4.11 mmol/L降低至1.34 mmol/L,表明突变后与底物亲和力增强。n值为Hill方程的希尔系数,代表协同性,其中n<1表示负协同反应;n=1表示非协同反应:n>1表示正协同反应,酶对配体的亲和力增大。AK的n值为1.71,突变体T379N/A380C/T65I/D173G的n值为1.07,n值降低,表明突变后AK的正协同性降低。
2.5 酶学性质表征
2.5.1 温度对AK活力的影响
如图5A所示,WT AK最适温度为25 ℃,突变后T379N/A380C/T65I/D173G AK的最适温度为30 ℃,最适温度提高5 ℃。在20~45 ℃温度范围内,突变体相对酶活力仍保持在60%以上,而且当温度低于20 ℃或高于26 ℃时,突变后的酶活力均高于WT,表明突变改善了酶的耐受温度性能,这对后期氨基酸发酵生产有益。
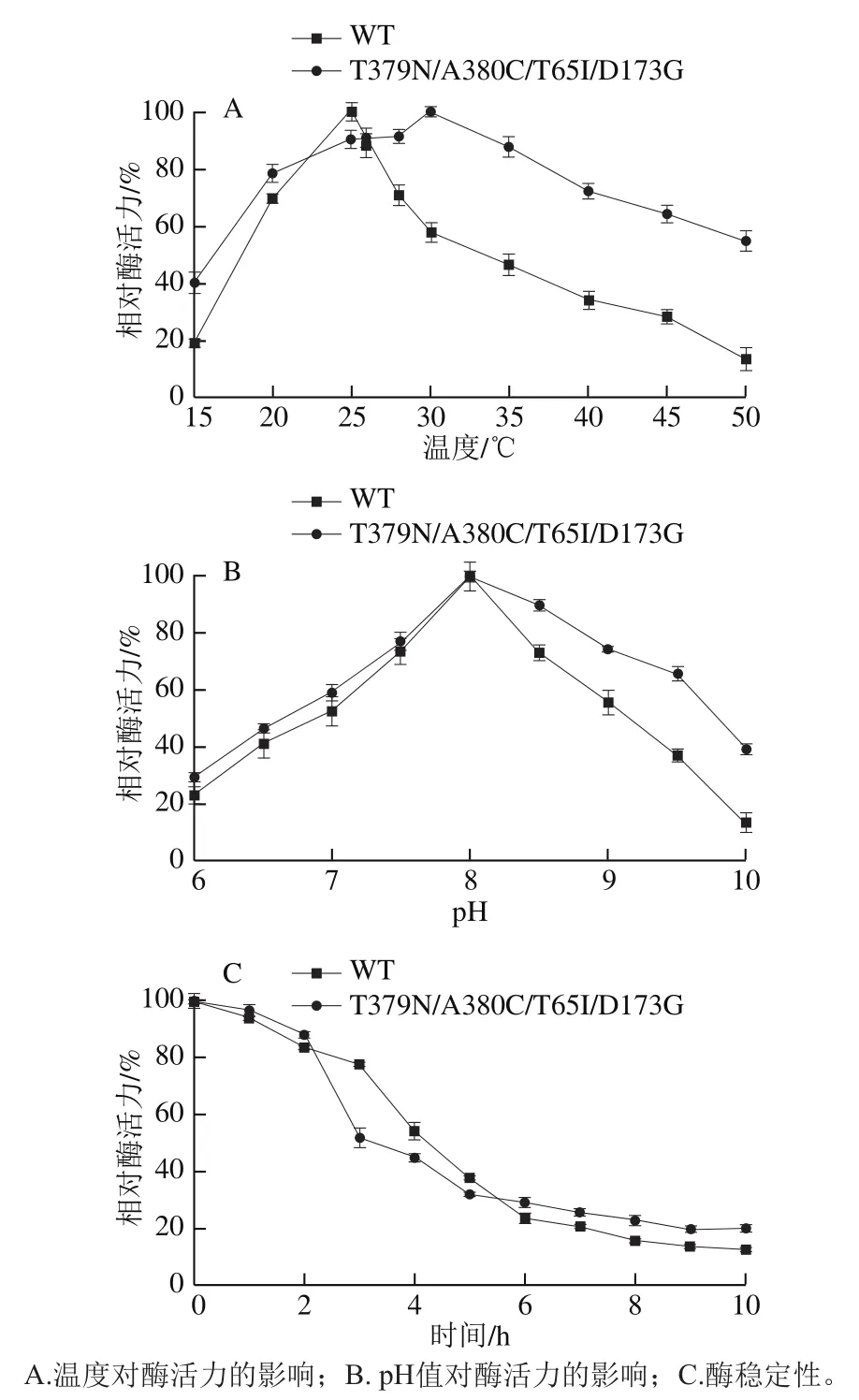
图5 WT和T379N/A380C/T65I/D173G的酶学性质表征Fig. 5 Enzymatic properties of WT and T379N/A380C/T65I/D173G
2.5.2 pH值对AK活力的影响
由图5B可知,WT和T379N/A380C/T65I/D173G的AK最适pH值均为8.0,然而,突变体T379N/A380C/T65I/D173G AK在pH 6~10条件下的相对酶活力均高于WT。
2.5.3 AK稳定性分析
分别在WT和T379N/A380C/T65I/D173G AK的最适温度和最适pH值条件下,测定酶稳定性,结果如图5C所示。计算得到WT AK的半衰期为4.24 h,而T379N/A380C/T65I/D173G AK的半衰期为3.56 h,较WT AK缩短了0.68 h,突变导致酶的稳定性减弱。在前2 h和6 h后,突变体T379N/A380C/T65I/D173G AK相对酶活力高于WT,10 h时突变体酶活力仍维持在20%左右,显著高于此时的WT。
2.5.4 底物抑制剂对AK活力的影响
根据表2可知,WT AK受Lys、Thr和Met抑制,抑制作用与抑制剂浓度呈正相关。单个抑制剂Lys浓度为10 mmol/L条件下,抑制率最高为56.22%;由于Lys和Thr两种抑制剂的共同作用,抑制效果最显著达到69.68%,WT AK的相对酶活力为30.32%。CpAK的抑制机制与CgAK相似,都是通过Thr和Lys的协同反馈抑制。当3种抑制剂共同作用时,最大抑制率为54.56%。突变体T379N/A380C/T65I/D173G AK在大多数抑制剂浓度下都有抑制作用减弱甚至被明显激活的趋势:在不同浓度Lys、Lys+Thr存在时都起到激活作用,尤其是在5、10 mmol/L的底物抑制剂Lys+Thr+Met条件下,相对酶活力分别达到114.30%和119.42%,说明突变体有效地解除反馈抑制,并且激活作用以剂量依赖性的方式增强。
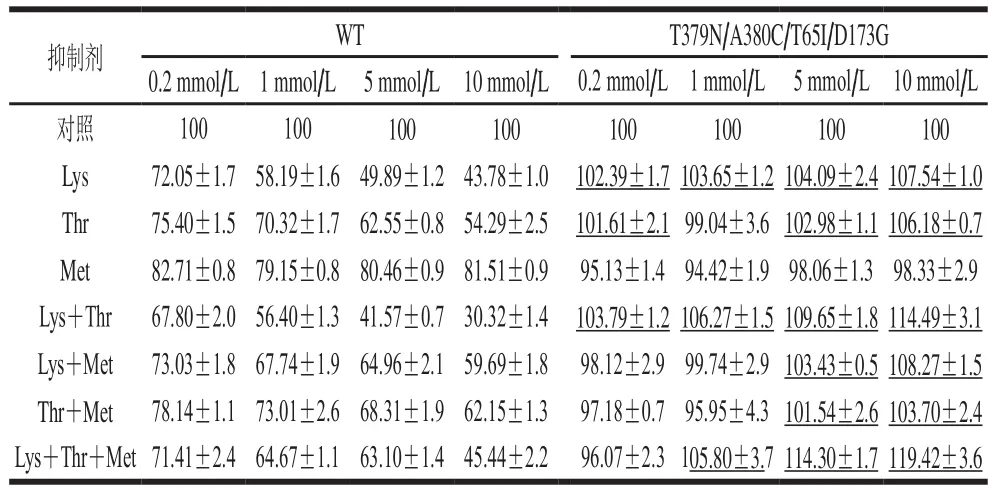
表2 底物抑制剂浓度对WT和T379N/A380C/T65I/D173G相对酶活力的影响Table 2 Effects of different concentrations of substrate inhibitors on the relative activity of WT and T379N/A380C/T65I/D173G%
2.6 工程菌构建及发酵测氨基酸含量
提取WT和构建成功的突变体T379N/A380C/T65I/D173G的质粒进行PCR扩增,产物进行1%琼脂糖核酸电泳验证,结果如图6A所示,1 000~2 000 bp之间有明显条带,与目的基因(1 317 bp)长度一致。线性化载体pEC-XK99E的大小为7 018 bp,与图6B条带位置一致。将胶回收的目的基因片段和线性化载体无缝克隆连接转入大肠杆菌感受态,然后电转化法转入北京棒杆菌中,质粒的核酸电泳验证结果如图6C所示,并测序证明WT和突变体T379N/A380C/T65I/D173G北京棒杆菌构建成功。
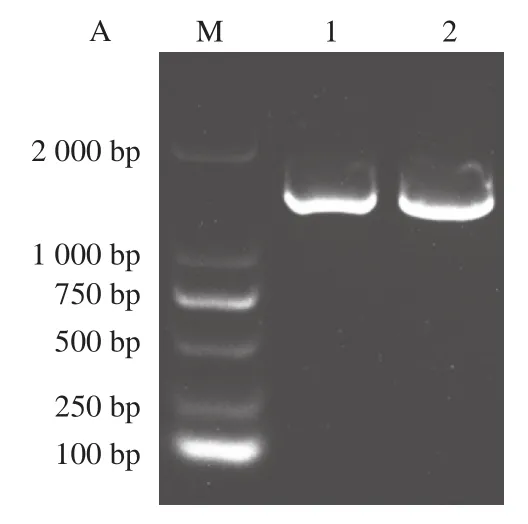
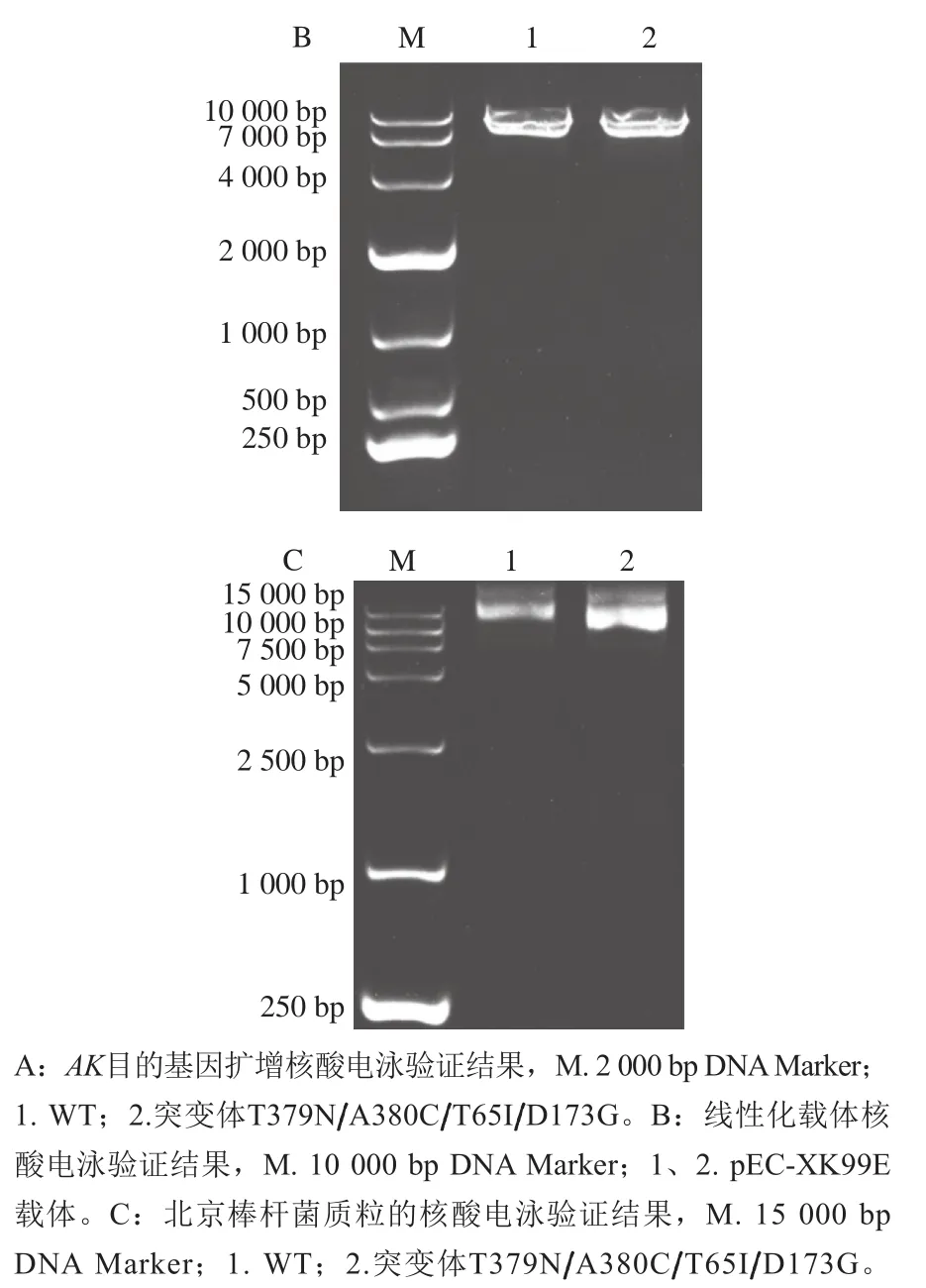
图6 工程菌构建的核酸电泳验证Fig. 6 Nucleic acid electrophoresis verification of constructed engineering strains
取30 h的发酵液进行氨基酸含量测定,结果如表3所示。相较于WT,下游氨基酸产量提高:Lys积累量由0.59 g/L提升至1.08 g/L,提高了83.05%;Thr产量由1.09 g/L增加至1.41 g/L,增加了29.36%;Met产量由0.13 g/L提高到0.17 g/L,提高了30.77%,Asp产量由0.33 g/L下降至0.15 g/L,下降了54.54%。原因可能是AK的Lys结合位点、ATP结合位点及底物结合位点通过突变,有效地解除了Thr和Lys和协同反馈抑制,提高了酶活性,使碳流量更多地进入天冬氨酸族氨基酸的合成途径,产量得以提高,然而突变后促进Asp流向三羧酸循环,从而使Asp积累量降低。
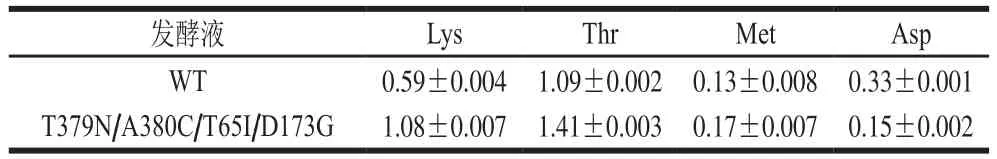
表3 发酵液中氨基酸产量Table 3 Amino acid contents in fermentation broth g/L
3 结 论
对关键位点Thr379、Aln380、Thr65、Asp173同时进行定点突变,成功筛选出获得酶活力显著提高到75.83 倍的AK突变体T379N/A380C/T65I/D173G,其与底物Asp的亲和力增强。酶学性质有利于菌株氨基酸发酵生产:最适反应温度由25 ℃提升至30 ℃,耐热性增强;最适pH值不变,酸碱耐受能力增强;半衰期由4.24 h缩短至3.23 h,稳定性略微减弱;有效地解除抑制剂的反馈抑制作用。此外,成功构建了北京棒杆菌工程菌,Lys产量达到1.08 g/L,Thr产量达到1.41 g/L,Met产量达到0.17 g/L,为优化Asp代谢途径和构建高产天冬氨酸族氨基酸提供参考。
