新生儿恶性常染色体隐性遗传骨硬化症一例报道
2022-01-27范佳英朱雪萍
范佳英,朱雪萍
(苏州大学附属儿童医院 新生儿科,江苏 苏州 215025)
骨硬化症(osteopetrosis)是一种极为罕见的、破骨细胞缺乏或功能缺陷导致骨吸收障碍的遗传代谢性骨病,其特征是骨质矿物质密度增加,但由于破骨细胞功能缺陷,易发生骨裂[1-3]。根据遗传类型可分为常染色体显性遗传骨硬化症(autosomal dominant osteopetrosis,ADO)、常染色体隐性遗传骨硬化症(autosomal recessive osteopetrosis,ARO)和X连锁骨硬化症3种亚型。其中婴儿ARO的平均发病率在1/200 000至1/300 000之间[4],主要表现为幼年起病,伴有严重且恶性的临床症状,临床具体表现为贫血、血小板减少、巨脑畸形、喂养困难、骨密度增高,一旦出现严重感染病死率极高。由于该病发病的不典型性,目前对该病的确诊主要依靠基因检测,治疗的主要手段为造血干细胞移植,但此方法仍需进一步探索。现将2018年苏州大学附属儿童医院新生儿科诊治的1例ARO患儿的临床资料报道如下。
1 病例资料
患儿男性,22日龄,因“发热2 d伴发现皮肤出血点1 d”入院。患儿入我院3 d前纳奶量减少,伴有哭闹不安,测体温(腋温)37.6 ℃,至当地医院住院治疗,查血常规示白细胞28.6×109L-1、C-反应蛋白(C-reactive protein,CRP)66 mg·dl-1,明显高于正常,血小板23×109L-1,诊断为“脓毒血症、新生儿肺炎、血小板减少性紫癜、尿路感染、心肌损害”,予“美罗培南40 mg·kg-14次·d-1抗感染、丙种球蛋白2.5 g 2次”提升血小板。入我院1 d前患儿颜面部出现散在出血点,体温仍有反复,阵发性哭闹,遂转诊至我院NICU。
既往史:无特殊。喂养史:患儿生后24 h内开奶,予母乳、普通足月配方奶混合喂养,奶量50~60 ml·次-1,8~10次·d-1,入我院3 d前患儿纳奶量减少,30~40 ml·次-1,6次·d-1。
出生史:患儿系G2P2,孕39周,2018年10月22日03时30分自然分娩娩出,出生体重3 750 g,生后Apgar评分不详,羊水性质、量不详,否认脐带绕颈,否认胎膜早破及羊水、胎粪吸入史。
家族史:父母亲均体健。本次妊娠为自然受孕,定期产检,母孕期曾有贫血(80 g·L-1),未予补充铁剂,孕母无血小板减少病史;G1为2017年9月出生的哥哥,3月龄时因“血小板减少、肺炎、脑发育异常(具体不详)”去世。
入院查体:体温37.3 ℃,脉搏140次·min-1,呼吸46次·min-1,体重 4.09 kg,血氧饱和度 96%(未吸氧下),身长52 cm,头围37.9 cm,胸围38 cm。神清,精神一般,刺激足底反应可,颜面部可及散在瘀点,按压不褪色,皮肤弹性可,稍干燥,前囟饱满,张力不高,心肺无特殊,腹软,肝脾肋下未及肿大,肠鸣音正常,四肢肌张力可,生理反射存在。
实验室检查:入院时血常规:白细胞18.05×109L-1,嗜酸粒细胞6.8%,网织红细胞百分比1.93%,血红蛋白115 g·L-1,血小板总数20×109L-1,中性粒细胞55.3%;超敏C反应蛋白87.32 mg· L-1;凝血常规:D二聚体>20 000 μg· L-1,活化部分凝血活酶时间49.8 s;降钙素原14.3 ng·ml-1;血清碱性磷酸酶518 U·L-1;自身抗体初筛:抗AMA-M2弱阳性(+)、抗Ro-52阳性(++)、抗SSA可疑(±);其余检查如肝肾功能、心肌三项等均未见异常。
影像学检查:胸片、头颅CT、MRI提示胸骨、颅骨骨质密度增高(图1);头颅CT示大脑半球脑白质密度减低;头颅MRI平扫示双侧颞叶及顶枕叶侧脑室边缘斑片状T1高信号影,胼胝体菲薄,透明隔间隙稍宽;心脏彩超示卵圆孔未闭。
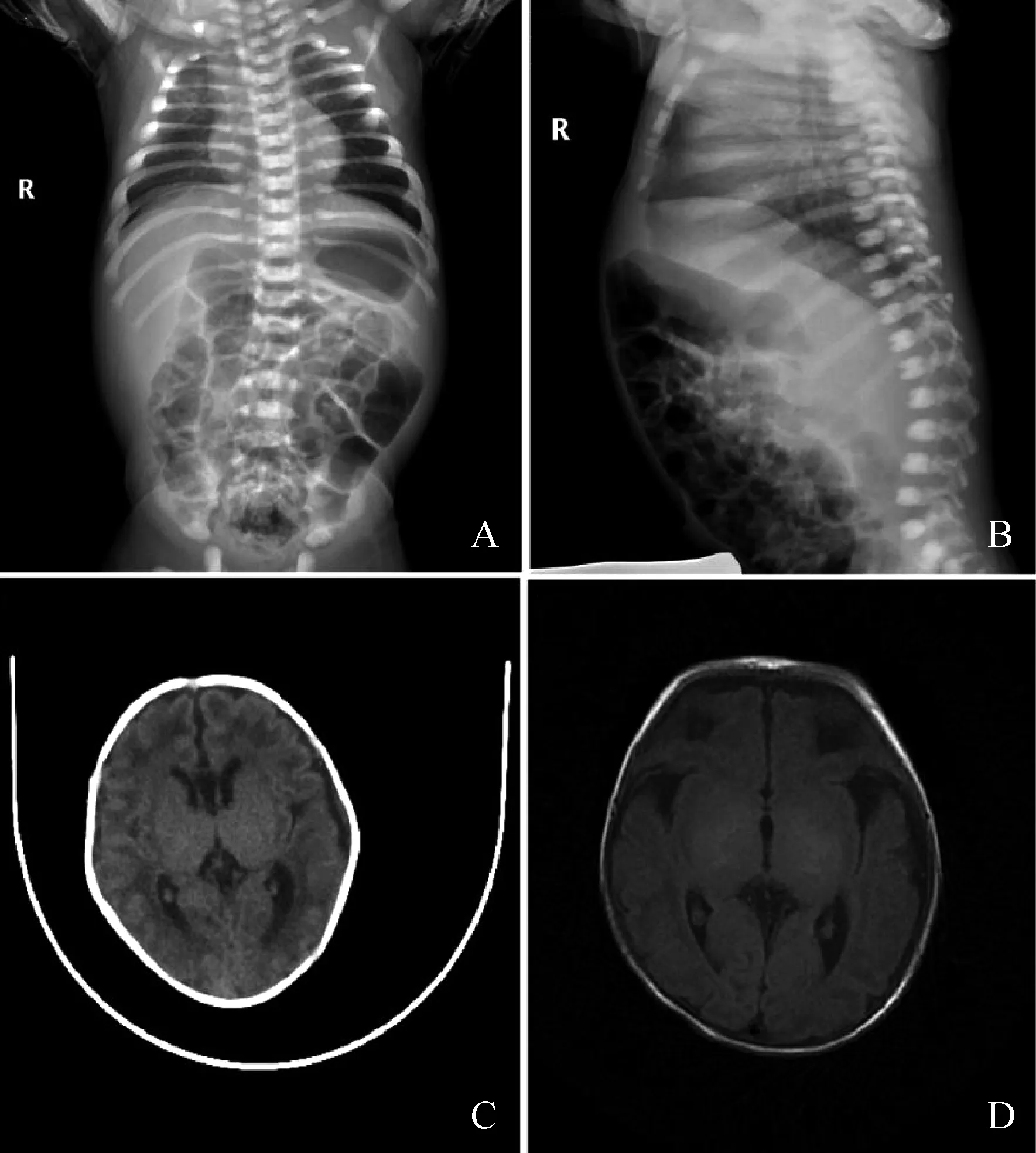
A、B分别为患儿胸片正侧位,提示胸骨骨质密度增高;C、D分别为头颅CT、MRI,均提示颅骨骨质密度增高
住院治疗经过及随访:患儿入院完善相关检查,予心电监护,美罗培南抗感染,卡络磺钠预防出血,入院查血小板总数20×109L-1,予滤白单采血小板1 U输注1次后血小板总数上升至128×109L-1,随后血小板波动在(54~88)×109L-1。入院第2天呼吸较费力,呼吸约60次·min-1,唇周稍有发绀,无发热,脉氧及血气分析未见明显异常,予头罩吸氧,并加用氟康唑预防真菌感染,3 d后气急好转,予停氧。结合病史及家族史,考虑基因病可能性大,完善基因检查。住院第18天改用头孢他啶抗感染。定期监测血常规,期间血红蛋白降至87 g·L-1,输注悬浮少白红细胞2次,血红蛋白上升至140 g·L-1。住院治疗第28天患儿出现腹泻,并有发热(热峰39.2 ℃),查轮状病毒阳性,予蒙脱石散口服,考虑感染反复,改用阿莫西林舒巴坦联合氟康唑抗感染,3 d后体温正常。住院第35天病情好转,予出院,出院时血小板总数74×109L-1。出院第2天基因结果回报,并告知家长。出院后电话随访,家属诉患儿未定期至门诊复查,出院半个月后精神一般,食纳欠佳,无发热,体重未见增长,未积极就医,于1个月后在家中去世。
分子遗传学:患儿父母签署知情同意书后,抽取患儿及其父母外周EDTA抗凝全血5 ml并提取基因组DNA,送至北京金准基因科技有限责任公司进行基因分析。经全基因组外显子测序(参考序列NM_001287.5),并对测序结果进行Sanger一代测序家系验证和qPCR验证,发现患儿CLCN7基因chr16:1506200存在c.824_830delTCTTCGA的杂合突变,同时该基因等位基因7-17 号外显子存在重复变异(图2)。
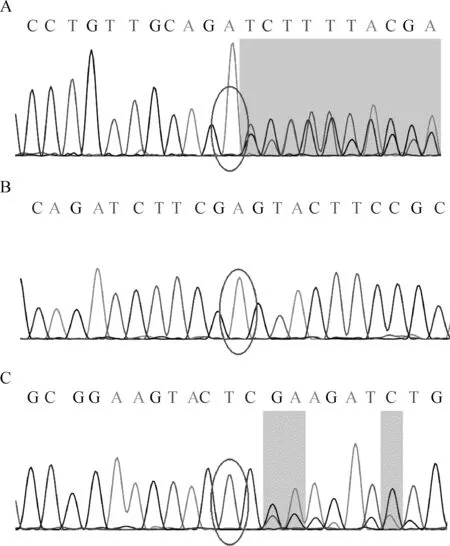
A.患儿以及患儿之父检测到chr16:1506200存在c.824_830delTCTTCGA的杂合突变,患儿之母不携带该杂合突变;B.患儿检测到CLCN7基因7-17外显子区域重复杂合突变;C.患 儿及其母亲均携带CLCN7基因大片段重复,患儿父亲不携带该重复突变
患儿chr16:1506200处7个碱基缺失c.824_830delTCTTCGA导致移码突变p.I275Sfs*47,属于恶性截断型突变,极有可能影响蛋白功能(PVS1);该突变在正常人群中未见携带,也未曾被报道为致病突变(PM2)。根据ACMG评级规则,该突变被归为疑似致病突变(likely pathogenic=PVS1+PM2)。同时,患儿7-17号外显子存在重复变异,极有可能影响蛋白的空间结构,进而影响蛋白的功能。一代家系验证发现患儿父亲CLCN7基因存在杂合移码突变c.824_830delTCTTCGA,即患儿的移码突变遗传自其父;患儿母亲CLCN7基因存在杂合重复变异,即患儿片段重复变异源于其母。CLCN7基因报道为常染色体隐性遗传或显性遗传,疾病遗传方式与突变位点类型相关,表型略有不同且常染色体隐性遗传患儿症状明显严重。患儿同时遗传了父母CLCN7基因的突变,且父母均未见疾病表型,符合常染色体隐性遗传,结合患儿脓毒血症、新生儿肺炎、血小板减少性紫癜、尿路感染、心肌损害、胸骨及颅骨骨质密度增高且婴儿期死亡等表型,确诊患儿为恶性常染色体隐形遗传骨硬化症(ARO)。
2 讨 论
ARO是婴儿期常染色体隐性骨骼疾病。TCIRG1、CLCN7和OSTM1基因是ARO的3个主要致病基因[5-8]。ARO预后差,大部分患儿在10岁之前因严重的贫血、出血或感染而死亡[9-11]。
我们对1例表现为发热、皮肤出血点的新生儿进行实验室检查及致病基因突变检测,结果显示患儿血小板低,胸部X线片具有骨质密度增高改变,患儿CLCN7基因携带新移码突变复合杂合大片段重复突变,两个突变分别遗传自患儿父母,且患儿父母均无疾病表征。
CLCN7基因位于第16条染色体,可编码长803个氨基酸的多肽链。该蛋白从127号位氨基酸开始直到597号位氨基酸的区间一共形成15个跨膜螺旋结构,这些跨膜螺旋共同组成了跨膜通道蛋白的功能性空间结构。加工成熟后的氯离子通道蛋白作为破骨细胞皱褶膜上的质子泵可将氢离子主动转运至破骨细胞内,提高胞内pH,进而降解破骨细胞吞饮的无机质,使得无机钙以离子的形式排入血液中,实现对骨形成以及骨质吸收的调控[3]。因此,CLCN7基因发生突变的患者会减少破骨细胞性骨吸收,从而引发骨硬化症。患儿1条等位基因存在移码突变c.824_830delTCTTCGA,导致氨基酸改变p.I275Sfs*47,即突变蛋白的多肽链仅前275位为正确的氨基酸序列,并在继续表达47位混乱序列后终止。突变蛋白缺失了10个跨膜螺旋结构,极有可能导致离子通道无法正确形成。另1条等位基因7-17外显子区域存在重复变异,导致199号氨基酸到539号氨基酸区间发生重复,即重复了10个跨膜螺旋结构,极可能使通道结构变形,影响氢离子的转运。两个突变均极有可能影响蛋白结构和功能,进而影响破骨细胞对骨形成和吸收的调节,导致骨硬化症的发生。
CLCN7突变可以引起3种类型的骨硬化症,包括常染色体隐性遗传骨硬化症ARO、中间型常染色体骨硬化症(intermediate autosomal recessive osteopetrosis,IAO)和Ⅱ型常染色体显性遗传骨硬化症(autosomal dominant osteopetrosis type Ⅱ,ADOⅡ),其中ARO通常发病于婴儿期,临床表现包括贫血、血小板减少、巨脑畸形、喂养困难、骨密度增高,一旦出现严重感染病死率极高。本例患儿根据新生儿期血小板减少、X线表现为骨密度增高,且婴儿期死亡,疾病症状严重等临床特征亦判断为ARO,进一步证明了患儿所携两个新突变的致病性。
对本例患儿双亲CLCN7基因的检测分析提示患儿父母均携带1个杂合的致病性突变,但两人表型均完全正常,这一结果提示虽然一些CLCN7基因突变可导致常染色体显性遗传的疾病,然而本例患儿两种新恶性致病突变均为常染色体隐性遗传。CLCN7 基因突变可引起婴儿恶性型、中间型、成人型骨硬化症[12]。其中成人型是常染色体显性遗传,由杂合突变造成,且通过对HGMD已记载致病突变的统计,我们发现恶行截断型突变占婴儿性常染色体隐性遗传骨硬化症的60%,占中间型常染色体隐性遗传的20.6%,却仅占导致成人型常染色体显性遗传骨硬化症突变的12.5%。这可能是由于截断型恶行突变对突变蛋白功能影响较大,导致纯合或复合杂合的患儿症状严重,而杂合携带者由于突变的等位基因无法表达功能性蛋白,突变蛋白在生成后大多被降解,刺激了正常等位基因的表达,使得体内正常的功能性蛋白维持在相当的水平;相反,普通错义突变虽然也影响了蛋白的功能,但是由于其整体结构改变不大,没有迅速被细胞自检机制清除,反而导致机体单倍体计量不足,表现部分骨硬化疾病表型。此外,某些修饰基因、表观遗传学修饰等因素也能使骨硬化症的表现型加重或减轻。
目前造血干细胞移植(hematopoietic stem cell transplantation,HSCT)是治疗骨硬化症的唯一有效措施[13]。Driessen 等[14]对122例HSCT治疗的ARO患儿进行长期跟踪随访,发现HLA相合患儿5年生存率可达73%。Orchard等[15]总结了1990—2011年65个移植中心193例婴幼儿骨硬化症患儿HSCT治疗情况,结果发现HLA相合患儿相较于其他供体移植的长期存活率更高,5年和10年生存率均为 62%。但并非所有骨硬化症患儿接受HSCT治疗后均能获得较好的疗效,大多数未经治疗ARO患儿将于10岁内死亡[16]。本例患儿亦未积极治疗,婴儿期死亡。
作者报道了1例复合杂合性突变导致ARO的案例,这种类型的病例在国内外已有报道[17-20],但患儿两条等位基因所携突变均为新致病突变。随着基因诊断的深入发展,新生儿疾病的早期诊断可以比较容易实现。新生儿疾病的形成是一个复杂的病理变化,因此其预后也会由这些变化的进展而决定。应综合评估患儿的一般情况,必要时积极完善基因检查,以早期明确诊断、早期治疗。
