均化在淫羊藿饮片质量均一性控制中的应用
2022-01-20王优杰刘敏臣张继全
薛 原 吴 梅 王优杰 冯 怡 刘敏臣 张继全
上海中医药大学中药现代制剂技术教育部工程研究中心,上海 201203
中药饮片的质量均一是保证制剂批间质量稳定的前提[1]。由于饮片质量受产地、基原、采收期等多种因素影响,批次间常存在较大质量差异,若以此为原料进行生产,质量波动势必传导至产品中,导致制剂批间质量均一性难以得到保证[2-3]。均化技术作为提高物料均一性的方法,已被运用于白酒勾调[4]、烟草加工[5]、水泥生产[6-7]等方面。为解决原料质量波动导致的制剂批间质量差异问题,国家药品审评中心于2020 年11 月发布了《中药均化研究技术指导原则(征求意见稿)》,旨在使用均化操作提高中药制剂批间质量均一性[8]。
淫羊藿是多基原药材,种属复杂多样[9-10]。研究表明,淫羊藿饮片的批间质量差异明显[9-10],这给制剂生产原料的选择及成品的质量稳定性控制带来了巨大挑战[11-12]。本研究以淫羊藿饮片为均化对象,以指标成分朝藿定C、淫羊藿苷的含量为基础设定均化目标,使用均化软件确定均化比例,对不同批次饮片进行均化,从指标成分含量及指纹图谱相似度的角度对均化结果进行评价,以期探索一种简单稳定的方法来提高中药饮片的质量均一性。
1 仪器与试药
1.1 仪器
Agilent 1260 型高效液相色谱仪(美国安捷伦公司);XP-205 型十万分之一电子分析天平、FA-2104 N 型万分之一电子分析天平(梅特勒-托利多仪器有限公司);SKT-210 HP 型超声波清洗器(上海科导超声仪器有限公司)。
1.2 试药
朝藿定C 对照品(批号:PS 000213,中国食品药品检定研究院);淫羊藿苷对照品(批号:110737-201516,中国食品药品检定研究院);水为超纯水;乙腈为色谱纯;其他试剂均为分析纯。
7 批淫羊藿饮片经上海中医药大学中药学院倪梁红老师鉴定,均为小檗科植物淫羊藿Epimedium brevicornu Maxim.的干燥叶,饮片相关信息见表1。
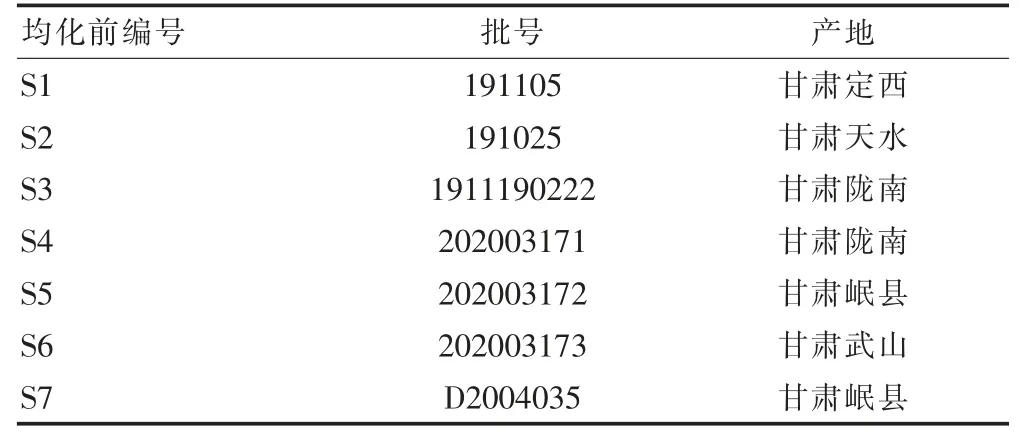
表1 饮片相关信息
2 方法与结果
2.1 对照品溶液的制备
分别精密称取朝藿定C、淫羊藿苷对照品适量,加甲醇制成浓度分别为1.16、0.98 mg/ml 的单一对照品母液。取1 ml 母液定容至25 ml 量瓶中,稀释得46.39、39.04 μg/ml 的单一对照品溶液,备用。
精密称取朝藿定C、淫羊藿苷对照品适量,加甲醇制成浓度分别为2.02、1.86 mg/ml 的混合对照品母液。取1 ml 母液定容至50 ml 量瓶中,稀释得40.38、37.23 μg/ml 的混合对照品溶液,备用。
2.2 供试品溶液的制备
取淫羊藿粉末(过三号筛)约0.2 g,精密称定,置具塞锥形瓶中,精密加入50%乙醇20 ml,称重,超声处理(功率400 W,频率50 kHz)1 h,放冷,再次称重,用50%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3 测定条件
2.3.1 指标成分含量测定色谱条件 色谱柱:Grace Apollo C18柱(250 mm × 4.6 mm,5 μm);流动相:乙腈-0.2%甲酸水溶液(26∶74);流速:1 ml/min;柱温:30℃;进样量:10 μl;检测波长:270 nm。
2.3.2 指纹图谱色谱条件 色谱柱:Pntulips QS C18Plus 柱(250 mm × 4.6 mm,5 μm);流动相:乙腈(A)-0.1%甲酸水溶液(B),梯度洗脱为0~35 min,26%A;35~55 min,26%~40%A;55~75 min,40%~55%A;流速:1 ml/min;柱温:30℃;进样量:10 μl;检测波长:270 nm。
2.4 确定均化比例
将均化前饮片的含量测定结果采用均化软件进行计算[13-14],以7 批淫羊藿饮片中朝藿定C、淫羊藿苷平均含量的120%为均化目标,设定目标值朝藿定C为0.85%、淫羊藿苷为0.84%,计算最优均化比例,得到6 批均化后饮片。均化比例见表2。

表2 饮片均化比例
2.5 方法学考察结果
2.5.1 系统适用性试验 精密吸取对照品溶液与供试品溶液各10 μl,按“2.3.1”项下条件测定,朝藿定C、淫羊藿苷色谱峰与相邻色谱峰分离度均>1.5,拖尾因子均为0.95~1.05,淫羊藿苷理论塔板数>8000。高效液相色谱图见图1。

图1 高效液相色谱图
2.5.2 线性关系 取“2.1”项下对照品溶液,加适量甲醇稀释得浓度为0.04、0.08、0.20、0.40、1.01、2.02 mg/ml的朝藿定C 对照品溶液;加适量甲醇稀释得浓度为0.04、0.07、0.19、0.37、0.93、1.86 mg/ml 的淫羊藿苷对照品溶液,按“2.3.1”项下测定。以朝藿定C、淫羊藿苷进样质量(mg)为横坐标,以峰面积为纵坐标,绘制标准曲线,计算回归方程及相关系数,见表3。结果显示,指标成分在一定质量范围内与峰面积线性关系良好。

表3 淫羊藿中指标成分的线性关系考察
2.5.3 精密度试验 精密吸取混合对照品溶液10 μl,按“2.3.1”项下连续进样6 次,计算峰面积相对标准偏差(RSD),结果朝藿定C 为0.32%、淫羊藿苷为0.34%,提示仪器的精密度良好。
2.5.4 稳定性试验 取S7 批供试品,按“2.2”项下制备供试品溶液,室温下放置,分别在0、2、4、8、12、24 h进样,按“2.3.1”项下测定。计算峰面积RSD,结果样品中朝藿定C 为0.85%、淫羊藿苷为0.26%,提示供试品溶液在24 h 内稳定。
2.5.5 重复性试验 取S7 批供试品,按“2.2”项下制备6 份供试品溶液,分别吸取10 μl,按“2.3.1”项下测定。计算峰面积RSD,结果测得朝藿定C 为1.73%、淫羊藿苷为1.27%,提示该方法重现性良好。
2.5.6 加样回收率 精密称定已知含量的S7 批供试品6 份,每份约0.1 g,分别加入一定量的对照品,按“2.2”项下制备供试品溶液,按“2.3.1”项下测定,计算朝藿定C、淫羊藿苷的平均回收率分别为99.75%、97.66%,RSD 分别为3.37%、4.33%,结果见表4~5。提示本方法回收率良好。

表4 朝藿定C 加样回收率结果(n=6)
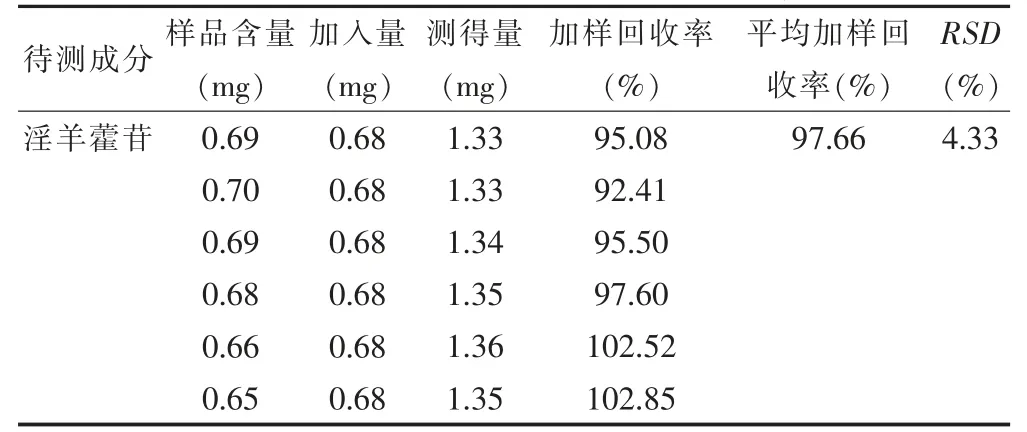
表5 淫羊藿苷加样回收率结果(n=6)
2.6 均化前后含量测定结果
以“1.2”项下7 批淫羊藿饮片作为均化前原料,以“2.4”项下得到的6 批为均化后原料,供试品溶液按“2.3.1”项下测定。计算得均化前后各成分含量及RSD,结果见表6~7。通过均化能使不同批次的淫羊藿饮片中朝藿定C、淫羊藿苷含量的RSD 明显降低,提示批间质量差异减小,均一性提高。

表6 均化前后朝藿定C 含量及RSD(%)
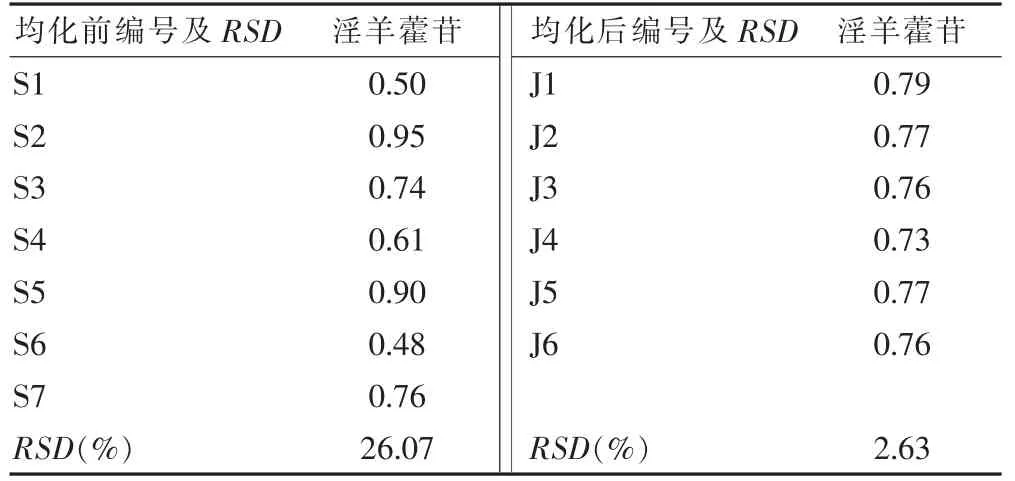
表7 均化前后淫羊藿苷含量及RSD(%)
2.7 指纹图谱研究结果
供试品溶液按“2.3.2”项下测定,采用《中药指纹图谱相似度评价系统2004A 版》进行数据处理[15-16]。设定S1 批为参照图谱,多点校正后自动匹配,得到对照指纹图谱及10 个共有特征峰。均化前后淫羊藿饮片的指纹图谱及对照图谱见图2~4,进一步计算得均化前后相似度结果见表8。均化前后淫羊藿饮片的相似度得以明显提高,提示不同批次质量基本达到均一稳定。
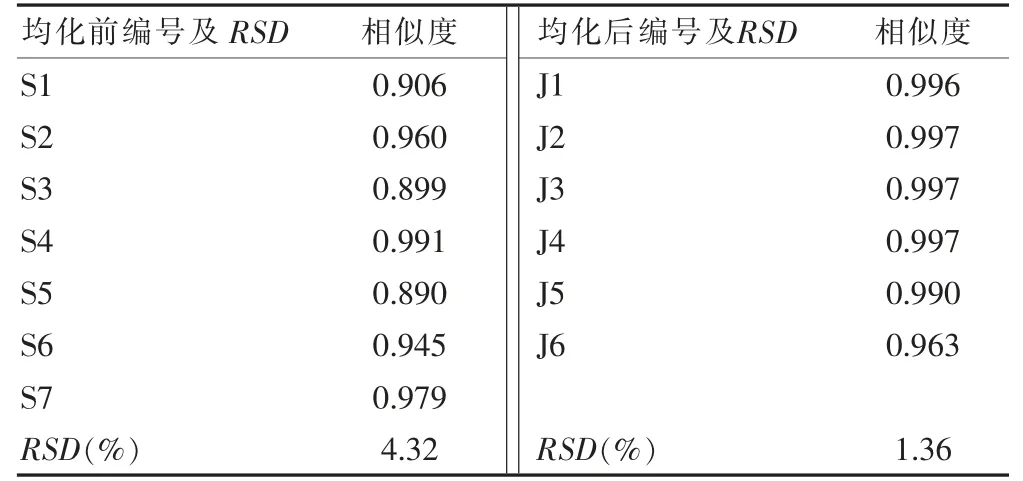
表8 均化前后饮片指纹图谱相似度及RSD

图2 淫羊藿饮片对照指纹图谱
3 讨论
本课题组前期研究发现同一产地不同批次的淫羊藿饮片质量差异较大,分析可能是受到土壤条件[17]、生长年限[18]、采收期[19]、产地加工方式[20]、炮制方法[21]等多种因素影响。不同产地的淫羊藿原料中,甘肃产地的饮片质量相对较为优良,合格率高[22-23]。

图3 均化前饮片指纹图谱

图4 均化后饮片指纹图谱
此外,淫羊藿中多糖类也为主要成分,若将其纳入考虑指标将更全面地反映淫羊藿饮片质量,但目前尚缺乏简单精确、误差小的测定方法,故本研究未将其作为均化评价指标[24]。同时,仅用相似度评价或许不能完全反映饮片质量及批次间的相似程度,在未来研究中还可以考虑纳入更多的指标,如峰面积、共有峰个数、峰面积和等[25]。
本研究方法中均化比例的确定借助软件,操作简单且精确度高,适合用于工业生产,值得推广。
