含活泼次甲基的烯基硼酸酯类化合物的设计与合成
2022-01-12蒋燕,唐攀,文龙,杨义
蒋 燕, 唐 攀, 文 龙, 杨 义
(四川轻化工大学 化学与环境工程学院,四川 自贡 643000)
有机合成是有机化学最重要的研究方向之一,其研究内容渗透到材料、生命、医药、农业、能源、环保等各个领域。例如,新药研发速度依赖先进化学合成技术快速、大量合成先导化合物、候选药物以及最终的药物分子[1];先进聚合物材料性能在很大程度上取决于所采用小分子单体的性质,除了采用新的聚合方法外,其每一次飞跃发展都与所用聚合单体的发展密切相关。因此,开发新型试剂以及有机合成子、发展新颖、高效合成技术,能够助推原始创新药物快速发展;同时为先进聚合物材料提供多样的单体[2]。
具有强亲核性的活泼亚甲基、活泼次甲基类化合物(1,3-二羰基化合物、2-吲哚酮类化合物、吡唑啉酮类化合物等)参与的亲核加成反应在过去20年内得到了广泛地研究,已经成为构建手性碳—碳、碳—杂键的重要合成方法[3-10]。有机硼试剂(芳基硼酸/硼酸酯、烯基硼酸/硼酸酯、硼酸盐等)由于其稳定性和多官能团容忍性,在金属催化的交叉偶联反应(Suzuki-Miyaura偶联[11-14]、Chan-Evans-Lam偶联[15-16])、无金属催化剂参与交叉偶联反应[17]、Hayashi-Miyaura反应[18-20]、Miyaura硼化反应[21]中有广泛应用。此外,有机硼试剂参与的反应被广泛应用于生物活性物质的后期修饰。
基于上述两个高活性且具有广泛反应活性的官能团,本文设计、合成了一类含有含活泼次甲基的烯基硼酸酯类化合物(Scheme 1)。首先,在氢氯二茂锆催化下实现频哪醇硼烷对3-溴丙炔的硼氢化加成反应,合成3-溴丙烯基频哪醇硼烷1,然后在碱的作用下1与氧化吲哚或β-酮酸酯进行亲核取代反应,合成了系列含活泼次甲基的烯基硼酸酯类化合物2和3,收率65%~89%,其结构经1H NMR,13C NMR和HR-MS表征。

Scheme 1

Scheme 2
1 实验部分
1.1 仪器与试剂
RY-2型显微熔点仪(温度未校正);Bruker-600 MHz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);MicroTMQ-TOF型高分辨质谱仪。
THF以二苯甲酮作指示剂,金属钠存在下在N2中蒸馏获得;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 3-溴丙烯基频哪醇硼烷(1)的合成
氮气保护下,在干燥的Schlenk瓶中加入氢氯二茂锆127 mg(0.5 mmol)和3-溴丙炔0.7 mL(8.1 mmol),搅拌下滴加频哪醇硼烷1.4 mL(9.6 mmol),滴毕,升温至60 ℃,反应10 h。反应液经硅胶柱层析(洗脱剂:石油醚)纯化得化合物1。
(2) 氧化吲哚基取代的烯基硼酸酯(2a~2d)的合成(以2a为例)
氮气保护下,在干燥的Schlenk瓶中加入2-吲哚酮0.133 g(1 mmol)和THF 5 mL, 冷却至-40 ℃,缓慢滴加正丁基锂溶液(2.5 M in Hexane)0.8 mL(2 mmol),滴毕,继续反应30 min;滴加10.25 g的THF(2 mL)溶液,缓慢升温至室温,反应2 h。反应完成后,将反应瓶置于冰水浴中,缓慢滴加饱和氯化铵溶液淬灭反应,用乙酸乙酯(3×5 mL)萃取,合并有机相,依次用饱和食盐水(20 mL)洗涤,无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4/1,V/V)纯化得化合物2a。
用类似方法合成2b~2d。
2a: 白色固体,收率76%;1H NMR(DMSO-d6)δ: 10.41(s, 1H), 7.14~7.19(m, 2H), 6.82~6.97(m, 1H), 6.81(d,J=7.5 Hz, 1H), 6.30~6.41(m, 1H), 5.36(d,J=17.9 Hz, 1H), 3.59(t,J=5.9 Hz, 1H), 2.75~2.82(m, 1H), 2.53~2.60(m, 1H), 1.15(s, 12H);13C NMRδ: 179.58, 149.48, 141.41, 129.45, 128.11, 124.76, 122.42, 109.80, 109.77, 83.36, 45.16, 37.03, 24.91, 24.88; HR-MS(ESI)m/z: Calcd for C17H22BNO3Na{[M+Na]+}322.1585, found 322.1584。
2b: 白色固体,收率65%;1H NMR(CDCl3)δ: 8.64(m, 1H), 7.14(t,J=8.0 Hz, 1H), 6.97(dd,J=8.2 Hz, 0.6 Hz, 1H), 6.79~6.74(m, 1H), 6.33(dt,J=17.8 Hz, 6.9 Hz, 1H), 5.51(d,J=17.8 Hz, 1H), 3.71~3.64(m, 1H), 3.20~3.10(m, 1H), 3.00(dddd,J=14.5 Hz, 6.7 Hz, 4.1 Hz, 1.3 Hz, 1H), 1.18(s, 12H);13C NMRδ: 178.70, 147.36, 143.10, 131.05, 129.53, 126.11, 123.18, 108.28, 108.23, 83.22, 46.06, 34.24, 24.81, 24.79; HR-MS(ESI)m/z: Calcd for C17H22BClNO3{[M+H]+}334.1376, found 334.1380。
2c: 白色固体,收率79%;1H NMR(CDCl3)δ: 8.57~8.73(m, 1H), 6.98(d,J=8.0 Hz, 1H), 6.88~6.95(m, 1H), 6.82(dd,J=8.4 Hz, 4.2 Hz, 1H), 6.62(ddd,J=17.8 Hz, 7.7 Hz, 5.6 Hz, 1H), 5.57(d,J=17.9 Hz, 1H), 3.55(dd,J=8.7 Hz, 4.5 Hz, 1H), 2.93~3.01(m, 1H), 2.52~2.63(m, 1H), 1.26(s, 12H);13C NMRδ: 179.50, 158.99(d,J=240.0 Hz), 148.82, 137.36, 130.93(d,J=8.0 Hz), 114.49(d,J=24.2 Hz), 112.74(d,J=24.8 Hz), 110.31, 110.26, 83.44, 45.57, 36.80, 24.88; HR-MS(ESI)m/z: Calcd for C17H22BFNO3{[M+H]+}318.1671, found 318.1675。
2d: 白色固体,收率70%;1H NMR(CDCl3)δ: 8.62(s, 1H), 7.13(dd,J=7.9 Hz, 1.7 Hz, 1H), 7.08(d,J=7.9 Hz, 1H), 7.05(s, 1H), 6.60(ddd,J=17.8 Hz, 7.6 Hz, 5.7 Hz, 1H), 5.55(d,J=17.9 Hz, 1H), 3.49(dd,J=8.5 Hz, 4.6 Hz, 1H), 3.01~2.91(m, 1H), 2.62~2.51(m, 1H), 1.26(s, 12H);13C NMRδ: 179.30, 148.83, 142.71, 128.24, 126.03, 125.34, 121.59, 113.23, 113.22, 83.44, 44.82, 36.80, 24.91, 24.89; HR-MS(ESI)m/z: Calcd for C17H21BBrNO3Na{[M+Na]+}400.0690, found 400.0691。
(3) 烯基硼酸酯3的合成(以3a为例)
氮气保护下,在干燥的Schlenk瓶中加入3-苯基-3-氧代丙酸甲酯0.178 g(1 mmol)和THF 5 mL,冷却至0 ℃,分批加入NaH 80 mg(2 mmol, 60%wt),继续反应20 min,再滴加10.25 g(1 mmol)的THF(2 mL)溶液,然后将反应液缓慢升温至室温反应过夜。反应完成后,将反应瓶置于冰水浴中,缓慢滴加饱和氯化铵淬灭,然后用乙酸乙酯(3×5 mL)萃取,合并有机相,用饱和食盐水(20 mL)洗涤,无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂,石油醚:乙酸乙酯=20/1,V/V)纯化得化合物3a。
用类似方法合成3b~3e。
3a: 微黄色黏稠液体,收率89%;1H NMR(DMSO-d6)δ: 8.01(dd,J=8.4 Hz, 1.2 Hz, 2H), 7.71~7.66(m, 1H), 7.56(dd,J=8.1 Hz, 7.5 Hz, 2H), 6.47(dt,J=17.9 Hz, 6.4 Hz, 1H), 5.35(d,J=18.0 Hz, 1H), 4.90(t,J=7.0 Hz, 1H), 3.58(s, 3H), 2.69(td,J=6.9 Hz, 1.4 Hz, 2H), 1.15(s, 12H);13C NMRδ: 195.05, 168.71, 149.76, 135.62, 133.95, 128.99, 128.66, 128.59, 82.86, 52.31, 51.43, 34.36, 24.57; HR-MS(ESI)m/z: Calcd for C19H25BO5Na{[M+Na]+}367.1687, found 367.1686。
3b: 微黄色黏稠液体,收率83%;1H NMR(CDCl3)δ: 7.89(d,J=8.2 Hz, 2H), 7.27(d,J=8.2 Hz, 2H), 6.60(dt,J=17.9 Hz, 6.4 Hz, 1H), 5.51(d,J=17.9 Hz, 1H), 4.42(dd,J=7.7 Hz, 6.6 Hz, 1H), 4.16~4.11(m, 2H), 2.87~2.80(m, 2H), 2.41(s, 3H), 1.24(s, 12H), 1.17(t,J=7.1 Hz, 3H);13C NMRδ:193.93, 169.55, 149.67, 144.64, 133.60, 129.55, 128.99, 83.31, 61.59, 53.22, 34.77, 24.88, 21.80, 14.14; HR-MS(ESI)m/z: Calcd for C21H29BO5Na{[M+Na]+}395.2000, found 395.2007。
3c: 微黄色黏稠液体,收率85%;1H NMR(DMSO-d6)δ: 7.98(d,J=8.9 Hz, 2H), 7.07(d,J=9.0 Hz, 2H), 6.47(dt,J=17.9 Hz, 6.4 Hz, 1H), 5.35(d,J=18.0 Hz, 1H), 4.77(t,J=7.1 Hz, 1H), 4.04(q,J=7.0 Hz, 2H), 3.85(s, 3H), 2.67~2.65(m, 2H), 1.16(s, 12H), 1.07(t,J=7.1 Hz, 3H);13C NMRδ: 193.09, 169.34, 163.63, 149.96, 131.02, 128.55, 114.13, 82.81, 60.79, 55.63, 51.39, 34.43, 24.54, 13.90; HR-MS(ESI)m/z: Calcd for C21H29BO6Na{[M+Na]+}411.1949, found 411.1954。
3d: 微黄色黏稠液体,收率78%;1H NMR(CDCl3)δ: 8.12(d,J=1.7 Hz, 1H), 7.90(d,J=7.9 Hz, 1H), 7.71(dd,J=8.0 Hz, 0.9 Hz, 1H), 7.36(t,J=7.9 Hz, 1H), 6.57(dt,J=17.9 Hz, 6.4 Hz, 1H), 5.50(d,J=17.9 Hz, 1H), 4.38(t,J=7.1 Hz, 1H), 4.15(qd,J=7.1 Hz, 5.0 Hz, 2H), 2.84(t,J=6.8 Hz, 2H), 1.24(s, 12H), 1.18(t,J=7.1 Hz, 3H);13C NMRδ: 193.04, 169.02, 149.11, 137.90, 136.54, 131.86, 130.41, 127.32, 123.24, 83.38, 61.86, 53.38, 34.56, 24.88, 14.12; HR-MS(ESI)m/z: Calcd for C20H26BBrO5Na{[M+Na]+}459.0949, found 459.0955。
3e: 微黄色黏稠液体,收率70%;1H NMR(CDCl3)δ: 7.61(d,J=1.0 Hz, 1H), 7.32~7.28(m, 1H), 6.61~6.55(m, 2H), 5.50(d,J=17.9 Hz, 1H), 4.23(t,J=7.3 Hz, 1H), 4.16(q,J=7.1 Hz, 2H), 2.87~2.80(m, 2H), 1.24(s, 12H), 1.19(t,J=7.1 Hz, 3H);13C NMRδ: 183.12, 169.08, 151.96, 149.30, 147.12, 118.72, 112.72, 83.32, 61.68, 53.48, 34.16, 24.88, 14.16; HR-MS(ESI)m/z: Calcd for C18H25BO6Na{[M+Na]+}371.1636, found 371.1643。
2 结果与讨论
2.1 合成
(1) 烯基硼酸酯逆合成分析
从烯基硼酸酯结构分析,其结构中的C—B键可以通过端炔与硼酸酯的硼氢化反应来构建,而活泼亚甲基上的侧链则可通过亲核取代反应引入。因此,拟通过容易获得的氧化吲哚、3-溴丙炔与频哪醇硼烷的反应来构建含有氧化吲哚取代的烯基硼酸酯类化合物(Scheme 2)。
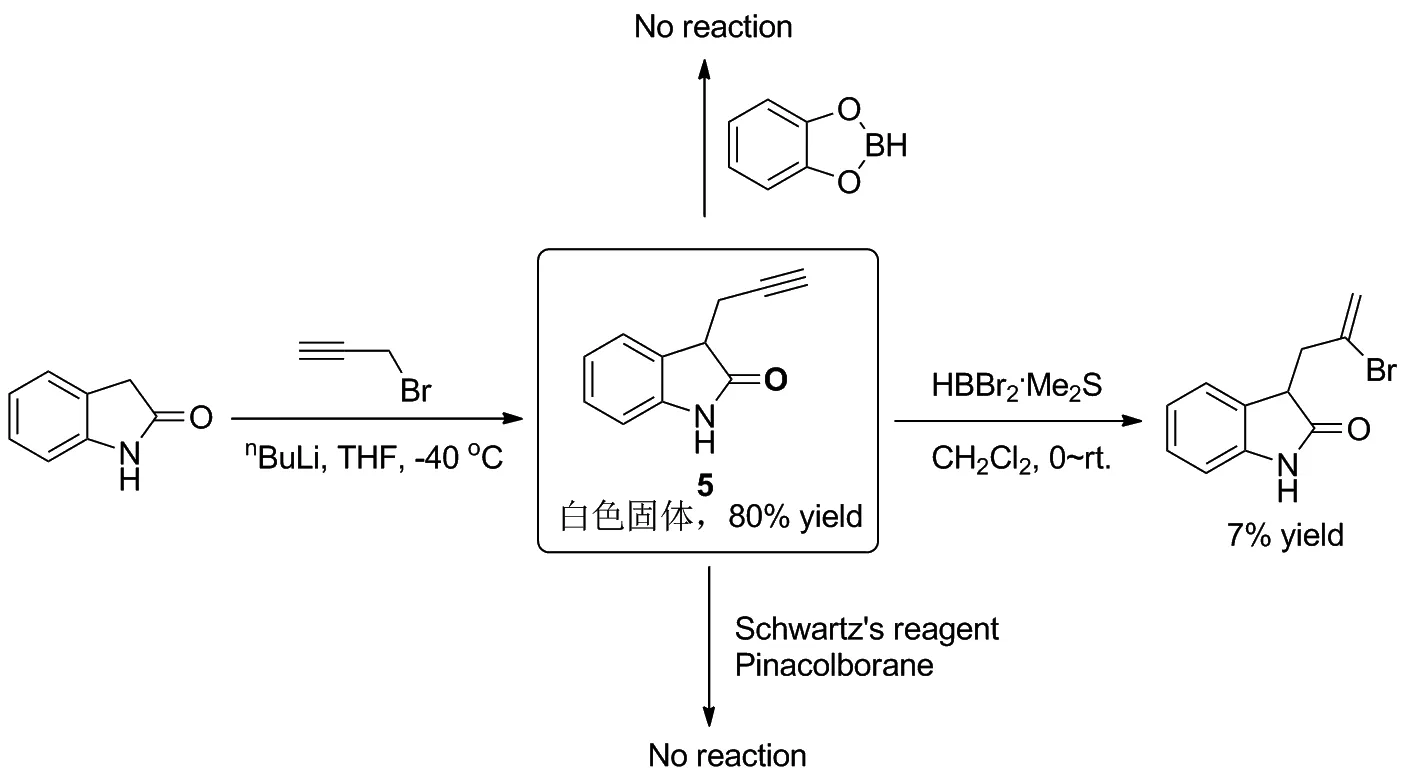
Scheme 3

Scheme 4
在此基础上,设计了两种合成路线。合成路线1通过在适当碱的作用下,首先在氧化吲哚3-位引入炔丙基侧链;然后再通过与合适的硼试剂发生端炔硼氢化反应而获得目标分子。合成路线2的策略为,首先将3-溴丙炔侧链转变成烯基硼酸酯侧链;然后在碱的作用下,在氧化吲哚3-位引入烯基硼酸酯侧链,从而获得目标化合物。
(2) 合成路线实施
根据合成路线1,选用正丁基锂作为碱,在-40 ℃下,成功地在氧化吲哚3-位引入炔丙基侧链,并获得80%的分离收率。随后,对端炔的硼氢化反应进行了一系列尝试。当选用二溴硼烷二甲硫醚作为硼试剂时,反应原料5大量存在,只分离得到少量HBr对端炔的加成产物。随后,尝试了儿茶酚硼烷作为硼氢化试剂,遗憾的是,反应未能进行。接下来,在氢氯二茂锆的催化下,尝试了5与频哪醇硼烷的反应,反应依然没有进行。实验结果表明,可能由于氧化吲哚刚性骨架的影响,端炔5不能发生硼氢化加成反应(Scheme 3)。
根据合成路线2,在氢氯二茂锆催化作用下,对3-溴丙炔进行硼氢化加成反应,以82%的分离收率获得3-溴丙烯基频哪醇硼烷1;然后在以正丁基锂作为碱,-40 ℃下,能够顺利地将烯基硼酸酯侧链连接在氧化吲哚3-位,以76%的分离收率获得目标分子(Scheme 4)。
在合成路线2的指导下,尝试了不同取代氧化吲哚与3-溴丙烯基频哪醇硼烷1的反应,均能有效合成目标分子。同时,以氢化钠为碱,可实现β-酮酸酯与3-溴丙烯基频哪醇硼烷1的反应,合成含有β-酮酸酯取代的烯基硼酸酯类化合物。
综上,先将3-溴丙炔转变成3-溴丙烯基硼酸酯侧链1,然后针对不同活泼亚甲基化合物的活性差异,选用不同碱,能实现多种含活泼次甲基的烯基硼酸酯类化合物的合成。
通过3-溴丙炔硼氢化加成反应,合成了3-溴丙烯基频哪醇硼烷中间体;然后在碱的作用下与氧化吲哚或β-酮酸酯进行亲核取代反应合成了9个结构新颖的含活泼亚甲基的烯基硼酸酯化合物,收率65%~89%。该类化合物可进一步作为有机合成子用于新型串联反应的开发。
