纤维素热解转化的研究进展
2022-01-05李承宇袁浩然王树荣
李承宇 ,张 军 ,袁浩然 ,王树荣 ,陈 勇
(1.中国科学院广州能源研究所,广东 广州 510640;2.广东省新能源和可再生能源研究开发与应用重点实验室,广东 广州 510640;3.中国科学院可再生能源重点实验室,广东 广州 510640;4.南方海洋科学与工程广东省实验室(广州),广东 广州511458;5.浙江大学 能源清洁利用国家重点实验室,浙江 杭州 310027)
当前,化石资源是石化工业中生产轻质烯烃和芳烃的主要原料来源。由于化石资源储量有限,生物质作为化石燃料碳源的替代品,具有储量丰富、可再生性强、不产生额外的二氧化碳排放等优点[1]。据估计,全球生物质年产量约为1000亿吨。国际能源署(IEA)预测到2035年,生物质能有望提供全球10%的主要能源供应,而到2050年,生物燃料可以替代世界上27%的交通运输所需燃料[2]。其中,木质纤维素类生物质由于来源广泛、价格低廉以及与食物链的非竞争性,成为一种极具吸引力的原料来源[3]。
目前,研究人员已经提出了多种生物质转化制备液体燃料的技术路径[4-6],其中,热解技术是在无氧或少量氧气存在环境下对生物质进行热分解,将其直接转化为高质量液体燃料和化学品的新技术,被认为是生物质资源化利用的最佳利用方式之一。热解产生的液体生物油是由水和数百种有机化合物组成的复杂混合物,如图1所示,其中,包含酸、醛、酮、醇、酯、脱水糖、呋喃、酚类以及部分低聚物等,这些热解产物可进一步高值转化用作液体燃料或者平台化学品[7-9]。

图1 纤维素热解制备各种高值化学品Figure 1 Various high-value chemicals from cellulose pyrolysis
纤维素作为木质纤维类生物质三大组分之一,占40%-60%[10]。因此,纤维素的热解行为可在很大程度上反映出木质纤维类生物质整体的热解规律,自20世纪50年代开始已经被用作木材和生物质热解的案例模型[11]。然而,由于纤维素氧含量较高,常规热解产生的粗生物油只能作为低等级、低热值的液体燃料,且产物分离和回收困难、成本高[12]。此外,热解生物油与化石燃料难以直接混溶,具有一定腐蚀性,在长期储存和运输过程化学性质不稳定,这些因素直接制约了纤维素热解技术的推广应用[13]。针对上述问题,近年来研究人员通过采用各种方式以期提升热解生物油的品质,如调控热解反应气氛、添加催化剂、产物减压蒸馏等[14-17]。
本综述将重点围绕纤维素热解方面的最新研究进展,系统介绍纤维素的结构特性、热解反应机理,并着重从常规热解和催化热解两个研究方向进行讨论。
1 纤维素的结构特性
纤维素晶体结构如图2所示,是由β-1→4糖苷键连接的D-吡喃葡萄糖单元组成的结晶聚合物,主要位于植物的细胞壁[18,19]。根据纤维素晶体的X射线衍射谱图,结晶重复单元是通过螺旋轴连接的两个脱水葡萄糖单元,左旋葡萄糖酐沿螺旋轴彼此旋转180°,因此,纤维素是以纤维二糖而不是葡萄糖为单体的线性均聚糖[20]。连接纤维素中的葡萄糖单元的糖苷键强度较低,倾向于在酸或高温条件下优先断裂。纤维素的化学式通常表示为(C6H10O5)n,其中,n为聚合度(DP)。天然纤维素的聚合度取决于其来源,通常来自木材的纤维素的聚合度为9000-10000,甚至可能高达15000[21]。在原纤维纤维素中,每个葡糖基单元有三个氢键,其中两个为分子内氢键,一个为分子间氢键。纤维素基质内大量的分子间氢键导致大多数纤维素分子的取向相近,从而形成微纤维。根据微纤维结构的有序程度,纤维素包含结晶(高度有序)区域和无定形(无规分布)区域,其沿着原纤维的取向周期性地或随机地分布,两种区域之间没有明显的分界,无定型区域比结晶区域更为活跃[22,23]。由于紧密排列的纤维素结构,纤维素结晶区域热稳定性比无定形区域更强。此外,分子内氢键可以稳定纤维素分子并抑制纤维素链的热膨胀,从而提高纤维的热稳定性。据报道,无定形纤维素的玻璃化转变温度为243-307 ℃,而纤维素晶体分解的最低温度高于300 ℃,因此,在热解过程中无定形区域首先发生降解,而结晶区域必须吸收足够的热量才能破坏氢键网络,促使晶体结构分解[3,24]。

图2 纤维素的晶体结构(虚线代表氢键)Figure 2 Crystal structure of cellulose(dotted lines represent hydrogen bonds)
纤维素中结晶区域的相对量一般采用结晶度指数进行评估,结晶度则可以通过样品XRD衍射峰计算获得。微晶纤维素的结晶度指数约为80%,生物质纤维的结晶度指数为30%-60%[3]。微晶尺寸是与纤维素晶体结构相关的另一个重要参数,指的是粉末形式的最小单晶。增加微晶尺寸使得非晶区域的晶体表面减少,导致结晶度指数增加[25]。结晶度指数和微晶尺寸都对纤维素热解行为有显著影响,近年来,研究人员已对其进行了广泛的研究。Kim等[25]对三种具有不同结晶度指数和微晶尺寸的天然纤维素样品进行热降解实验,研究发现随着结晶度指数和微晶尺寸的增加,DTA和DTG曲线中的初始热分解温度和峰值温度向高值移动,该现象主要是纤维素热分解过程中样品结晶区域传热速率差异导致的。
除结晶度指数和微晶尺寸外,结晶同质异形体是纤维素晶体结构的另一个关键参数。纤维素大分子中的羟基相互作用形成大量分子内和分子间氢键,产生各种有序的晶体排列,从而形成不同的结晶同质异形体(图3)。通过对其特征XRD谱图和固态13C NMR核磁图谱分析,可以识别出几种结晶同质异形体,其中,纤维素Ⅰ、Ⅱ和Ⅲ是最典型的同质异形体[26]。纤维素Ⅰ是植物细胞壁中纤维素的天然形式,广泛存在于自然界中,具有典型的二维层内氢键网络,其他形式的纤维素均可由纤维素Ⅰ改性制备。Vanderhart等[27]在研究中发现纤维素Ⅰ包含了纤维素Ⅰα和纤维素Ⅰβ这两种亚变体结构。纤维素Ⅰα在藻类和细菌纤维素中普遍存在,纤维素Ⅰβ则主要存在于高等植物的纤维素中[28]。纤维素Ⅰα可以通过热处理转化为热力学更稳定的纤维素Ⅰβ,因此,纤维素Ⅰα也被称为纤维素Ⅰβ的亚稳态相[29]。纤维素Ⅱ可以通过再生(溶解和重结晶)和碱处理两种方式从纤维素Ⅰ中制备,具有层内和层间氢键三维网络组成的反平行片状结构。通过对纤维素Ⅰ或纤维素Ⅱ进行无水氨处理则可以获得纤维素Ⅲ,其具有类似纤维素Ⅱ的层内和层间氢键,但保持与纤维素Ⅰ一致的平行链结构[30]。结晶同质异形体的类型亦对纤维素热解过程具有一定影响。纤维素Ⅰ热解生物油的主要成分包括糠醛(FF)和1,4:3,6-二脱水-α-D-吡喃葡萄糖(DGP),纤维素Ⅱ和无定形纤维素的主要产物由糠醛(FF)和5-羟甲基糠醛(5-HMF)组成,而且纤维素Ⅱ和无定形纤维素中氧原子主要以CO2形式除去[31]。

图3 纤维素Ⅰ、Ⅱ和Ⅲ的结构模型[30]Figure 3 Molecular models of cellulose allomorphs[30](with permission from ACS Publications)
此外,结晶度也是影响纤维素Ⅰ和纤维素Ⅱ热解产物分布的重要因素之一。对于纤维素Ⅰ,低结晶度促进了纤维素的脱水反应和水溶性反应中间体中低聚糖的生成,并有利于呋喃的生成,主要产物左旋葡聚糖(1,6-脱水-β-D-吡喃葡萄糖,LG)的选择性则随着结晶度降低而逐步减小。只有高结晶度的纤维素Ⅱ样品才能产生由LG主导的热解产物,而纤维素Ⅲ热解产物组成则与原料结晶度无直接关联[30,32]。同时,高结晶度纤维素Ⅰ、Ⅱ以及各种结晶度的纤维素Ⅲ样品均具有最高的起始失重温度和最低的焦炭产率[30]。研究人员还观察到无论相对结晶度如何变化,纤维素Ⅲ样品在热解时都表现出明显的熔融相变,而高结晶度的纤维素Ⅰ在热解气化过程中却没有发生实质的相变[30]。
2 纤维素的常规热解
2.1 纤维素热解机理
在纤维素加热处理过程中,首先聚合物结构中化学键被破坏,引起挥发性物质的释放和残余基质内发生重排反应,上述初级转化反应被认为是纤维素热解转化的主要机制[33]。在初级产物形成后,由于一些挥发性化合物在该反应体系化学性质不稳定,可以进行额外的转化,即二次反应,二次反应对于最终热解产物的形成具有重要意义[34]。
不同木质纤维类生物质的初级转化具有共同的特征,均包含三种主要转化途径,即焦炭的形成、解聚和碎片化(图4)[33-37]。其中,焦炭是生物质转化形成的固体残余物,具有芳族多环结构。焦炭形成过程容易发生分子内和分子间重排,提高了产物结构的网状化程度和热稳定性。该途径主要包括苯环的形成和芳环在多环结构中的组合,而且这些重排反应通常伴随着水或不凝气体的生成[33,37,38]。解聚指的是破坏聚合物内单体之间连接的化学键,并在断裂后形成的新链端发生稳定化反应。解聚反应会持续进行,导致聚合物聚合度不断降低,直到转化为易挥发的小分子从体系中分离出来,一般会形成衍生单体、二聚体、三聚体等常温下为液态的产物[36]。碎片化则是高温下聚合物内大量共价键断裂,甚至单体单元内部结构共价键直接断裂,从而生成链状小分子,如酸、醛、醇和一些不凝气体等[33]。

图4 生物质转化的主要机制途径(温度从左到右逐渐升高)Figure 4 Main mechanism of biomass pyrolysis
二次反应具体指的是挥发性产物中不稳定部分发生的裂解或重组反应。裂解是挥发性化合物分子内化学键断裂变为小分子的过程,由于这些化学键在聚合物内也可以通过碎片化进行断裂,因此,裂解产物和初级转化中的碎片化产物具有相似性[39]。重组则是挥发性的物质互相组合,从而产生更高分子量的化合物,甚至会形成在反应温度下无法挥发的固体。通过气相产物分析,多环芳烃(PAH)的存在说明反应条件有助于重组反应的进行。二次反应通常在焦炭表面、反应器表面或催化剂上进行,其中,重组过程会在反应物内形成二次焦炭,或者沉积在催化剂表面导致催化性能下降[40]。
2.2 纤维素热解机理的研究进展
自上世纪70年代起,有关纤维素热解机理的研究就已取得了较显著的进展,主要侧重于纤维素糖苷键断裂、CO、CO2等气态产物和焦炭的形成,液体产物形成等方面。这些机理研究形成了目前纤维素甚至生物质热解的理论基础,其中,Anca-couce的研究工作最具代表性[41]。
2.2.1 Broido-Shafizadeh纤维素热解模型(1979年)
该模型最初由 Kilker和Broido提出[42],后来由Shafizadeh和Bradbury优化得到[43]。首先,纤维素的解聚产生活性纤维素,降低了聚合度,但不释放任何挥发物。活性纤维素具有较低的聚合度,可以通过两种竞争性反应分解:一是产生焦炭和永久性气体的缓慢反应;二是产生左旋葡聚糖(LG)和其他脱水糖类的热解生物油的反式糖基化解聚等反应。当温度高于300 ℃,有助于热解油的生成。
在该模型中,葡萄糖分解产物可以通过热解油二次脱水反应在一次热解后形成,由此产生呋喃、吡喃类化合物或其他糖类。这些脱水产物也是形成二次焦炭的中间化合物。糖单元的裂解产生各种羰基化合物和烯烃,以及水、CO、CO2和焦炭。研究表明,上述裂解反应产物与纯LG热解产物非常接近。此外,产生焦炭和气体的反应活化能低于产生热解油的反应,因此,在低温下有利于一次焦炭的形成[42,43]。
2.2.2 Piskorz纤维素热解模型(1989年)
随后研究表明,部分环裂解产物(碎裂产物),如羟基乙醛(HAA)等低分子量化合物(LMWC),尤其是羰基化合物,并不是通过LG或其他糖类的二次转化反应形成,而是由活性纤维素直接形成的。在Piskorz等提出的新模型中,初始阶段涉及低温下焦炭形成和纤维素聚合程度迅速降低产生活性纤维素之间的相互竞争。当聚合程度充分降低后,环碎裂之间和反式糖基化解聚之间存在竞争关系,前者产生羟基乙醛等LMWC,而后者生成LG和其他糖类[44]。
2.2.3 Banyasz纤维素热解模型(2001年)
Banyasz等后来又对Piskorz模型进一步优化[45,46]。在保留低温炭化反应的同时,认为活性纤维素先转化为液态LG等热解油成分和LMWC中间体,两种产物为竞争关系。液态焦油蒸发后发生二次炭化生成CO2和焦炭,而LMWC中间体主要发生两种互相竞争反应,一种生成HAA,另一种生成甲醛和CO。值得注意的是,该模型中CO和CO2是通过两种不同途径生成的。Banyasz等还计算了环裂解生成LMWC中间体和反式糖基化生成LG等热解油的活化能,其中,环裂解反应所需的活化能更高,因此,在高温下优先于反式糖基化反应[46]。
2.3 热解产物的影响因素
由前述纤维素热解机制可以看出,纤维素常规热解产物分布主要由热解温度决定。根据热解温度的不同,纤维素的热解产物组成会有明显的差异。下面将分别介绍不同温度区间纤维素主要热解反应产物。
2.3.1 形成活性纤维素或脱水纤维素(150-300 ℃)
当热解温度较低时,纤维素的质量损失主要由脱水反应造成。在此温度区间,纤维素分解比较缓慢,但会发生一些结构变化。如图5所示,主要包括部分解聚生成活性纤维素、聚合度降低、还原端基团脱水反应、非结晶分子的结晶等[47]。在此温度区间,纤维素热解最终生成焦炭和其他固体混合产物,基本不会产生CO、CO2和其他有机热解气体[48]。因此,较低温度热解不利于生物油或热解气的合成,但是可以获得聚合度较低、反应活性较高的固体产物,有利于后续转化。

图5 较低温度(150-300 ℃)纤维素的结构变化Figure 5 Structural changes of cellulose at lower pyrolysis temperatures (150-300 °C)
2.3.2 纤维素链的解聚(300-380 ℃)
当热解温度高于300 ℃时,纤维素结构中的糖苷键极其活泼,导致很多反应同时发生,产生大量挥发性产物和部分焦炭[18,34]。挥发性产物包括脱水糖、呋喃化合物,以及通过解聚、脱水和片段化反应形成的C1-C3片段。如图6所示,最主要的标志性产物就是左旋葡聚糖(LG),最高产率可以达到60%左右[48,49]。除此之外,少部分呋喃糖衍生物 1,6-脱水-β-D-呋喃葡萄糖(AGF)随之产生,而且具有额外分子内醚键的脱水糖如1,4:3,6-二脱水-α-D-吡喃葡萄糖(DGP)也被检测出来[18]。同时,通过脱水反应还会产生各种呋喃和吡喃化合物,主要包括FF和5-HMF等。此外,小分子碎片产品中醛(HAA、甲醛、丙烯醛和乙二醛)、酮(羟基丙酮)和酸(乙酸和甲酸)在最终产物中占比较大[50,51]。由此可见,在纤维素常规热解中,生物油主要在300 ℃以上开始形成,而随着温度的进一步升高,纤维素解聚加剧,产物中生物油比例也随之升高。

图6 纤维素热解形成主要挥发性产物左旋葡聚糖和糠醛的机理Figure 6 Mechanism of cellulose pyrolysis for producing volatile products (LG and FF)
2.3.3 纤维素高温下的炭化过程(380-800 ℃)
当热解温度为380-800 ℃时,纤维素炭的芳族多环结构发生分子内和分子间重排反应,逐渐向网状化程度和热稳定性更高的类似木质素炭结构转化,同时产生部分热解气体(甲烷、氢气、CO)(图7)[52,53]。其中,甲烷的生成温度为500-600 ℃,在此温度区间芳族多环上的甲基残基浓度降低,可推测甲烷是由多环芳烃之间发生去甲基化反应生成。CO和氢气的生成方式与甲烷相似,其转化温度为500-800 ℃[54]。当温度高于600 ℃时,二次反应中的裂化反应对产物分布影响较为显著,而800 ℃以上的热解温度则有利于重组反应的发生,产生少量PAH等。此外,热解产生的挥发性气体在反应器内停留时间越长,二次反应就越容易发生[40,55]。综上所述,在热解温度达到500 ℃以后,随着温度升高,热解气体产物占总产物的比例增大,而生物油的产率由于重组反应的加剧而有一定程度的下降。因此,高温有利于气化产物和焦炭的形成。

图7 苯环取代基团转化生成CH4、CO、H2和焦炭Figure 7 Conversion of substituent group from benzene ring to CH4, CO, H2 and char
2.3.4 其他影响纤维素热解产物分布的因素
热解温度对纤维素热解产物组成具有重要影响,与此同时,升温速率和原料粒径的调控对热解产物分布也起到关键作用。当热解过程升温速率较低时,纤维素结构内最弱的化学键发生断裂,但是其他化学键依然保持稳定,此时纤维素结构只受到轻微影响,部分重排反应的发生导致基质变得更加稳定,最终抑制挥发性化合物的生成。而当升温速率增加至临界值时,不同类型的化学键几乎同时被破坏,挥发性化合物在重排之前就会被释放出来[56,57]。粒径对反应产物的影响主要体现在粒径与原料内部的加热速率直接相关,粒径较大的原料内部升温速率较慢,在升温的过程中初级热解的前沿会从颗粒外表面向中心逐渐移动。因此,随着热解反应的进行,原料颗粒内会形成不同的区域(图8):外部区域挥发性物质完全释放形成的焦化层、正在释放挥发性物质的中间区域和尚未发生热解的内部原料区域。因此,初级热解产生的物质必须经过多孔炭孔道结构才能离开颗粒,进而增加了挥发性气体间的均相反应、挥发性气体与焦炭之间的非均相反应等二次反应的可能性。因此,在原料粒度较大时,其热解液体产物产率显著降低,而轻质气体与焦炭的产率会有所提高[56-58]。

图8 纤维素颗粒热解过程剖面示意图Figure 8 Schematic cross-section for the pyrolysis of cellulose granules
2.4 纤维素常规热解产物分析方法
为了研究热解产物的产率和组成,研究人员已经开发出各种类型反应器研究生物质热解特性,包括固定床反应器、流化床反应器、烧蚀反应器、旋转锥反应器、螺旋钻反应器和真空反应器等[3,34]。反应器的选择取决于能影响到反应进程的主要参数,如原料粒度、传热速率、固体停留时间和挥发物等[34]。常规气态馏分(即CO2、CO、甲烷和氢气等)的组成一般采用炼厂气气相色谱进行定性定量分析。对于热解油,其中水分含量通常采用Karl-Fischer滴定法测定,采用气相色谱与质谱联用分析主要的有机化合物组成[34]。对热解机理的深入理解需要掌握关于热解过程更详细信息,包括热解产物定量、反应物和产物演变过程以及反应中间体的捕获分析。为了实现上述目标,一些更为先进的研究手段被应用在纤维素热解过程中,主要包括裂解器-气相色谱-质谱联用/火焰离子化检测器(Py-GC-MS/FID)、热重-质谱/热重-傅里叶变换红外光谱联用(TG-MS/TG-FTIR)、原位光谱(2D-PCIS)、同位素标记和原位电子顺磁共振光谱(EPR)、多点式激光光谱(PIMS)等[34]。
对于Py-GC-MS/FID,微量热解器结合气相色谱(GC)、质谱(MS)和火焰离子化检测器(FID)可以准确地定性定量分析生物质热解产物。在微量热解器中产生的热解蒸气可以直接输送至GCMS/FID进行分析,无需在热解过程冷凝收集产物。经过GC分离之后,可以通过MS检测器定性地确定热解蒸气的组分,结合FID或其他类型检测器可进一步进行定量分析。与常规实验室规模反应器中停留时间相比较,挥发物在微型热解器中的停留时间可缩短至15-20 ms[59],因而可有效地抑制二次反应的发生,因此,Py-GC-MS/FID成为当前研究生物质热解反应的主要技术手段[3]。
由于Py-GC-MS/FID只能进行非连续分析,不能获得随时间或温度变化的中间产物演变特征信息[3]。基于这一问题,研究人员通过耦合TG与MS或傅里叶变换红外光谱(FT-IR)以获得样品升温过程热解产物组成与演变机制。这两种技术的开发可以有效监测生物质热解挥发分的变化,记录质量损失过程,并确定主要的挥发性物种及对应的温度范围。同时,鉴于解聚产物结构与热解残余物中一个单元非常接近,这些耦合还有助于理解样品未挥发部分组成的演变规律。在热解产物研究方面,则提供了热解转化初始阶段纤维素不稳定结构碎裂和炭化过程中重排反应的温度信息。TG-FTIR技术不能测定同源双原子种类,对具有相似功能基团的化合物难以区分,而TG-MS则可通过测量挥发物的质荷比(m/z)解决这个问题[60]。然而,由于生物质热解产生的挥发物成分复杂,对高分子量化合物采用TG-MS技术进行分析仍具有局限性。目前,TG-MS技术主要用于测定纤维素热解过程中轻质气体的释放行为,如H2、CH4、H2O、C2H4、CO和 CO2[61-64]等。为了解决单独使用TG-MS或TG-FTIR技术的不足,已开发出TG-MS-FTIR三联用技术。此外,通过FT-IR、XRD或NMR分析,也可以得到更多有关不同温度下热解过程固体残余物发生化学变化的信息。
Py-GC-MS/FID和 TG-FTIR(TG-MS)技术均侧重于热解最终产物的分析,无法提供有关反应过程的详细信息。为了可以直接反映出热解反应过程,研究人员开发了原位光谱学分析技术。原位FT-IR是最常见的光谱分析手段,可用于表征热解过程生物质表面官能团的变化[65]。其他原位光谱技术也相继被开发用于研究热解过程,例如原位同步辐射X射线衍射[66]、时空解析粒子的漫反射原位光谱(STR-DRiSP)[58]和原位1H NMR光谱[67]等。具体地,原位同步辐射X射线衍射主要用于研究微晶结构的降解过程,STR-DRiSP可以在高空间(10 μm)和时间(1 ms)分辨率快速热解过程中测量生物质组成,原位1H NMR光谱主要用于研究生物质热解过程中的质子迁移率。Noda等[68]在2D NMR技术基础上开发出广义二维微扰相关红外光谱(2D-PCIS),该技术能够破译特定键中的扰动诱导变化,主要用于研究生物质炭形成或烘焙过程中官能团的变化。除了原位光谱学之外,同位素标记法和电子顺磁共振(EPR)光谱等也能通过确定每种热解产物中特定原子的来源或是通过检测自由基以研究纤维素的热解机制[3]。
2.5 纤维素热解的动力学参数
热重分析(TGA)是一种快速有效的定量方法,用于掌握非等温和等温条件下的热解反应,确定纤维素热分解的动力学参数[69]。这些动力学参数包括活化能(Ea)、反应阶数(n)和指前因子(A)。
活化能是提供有关纤维素热解性能相关信息的主要因素。通过 TGA确定动力学参数对于设计和建立有效、安全和合理的热解反应进程至关重要[70]。Kissinger、Ozawa、Flynn-Wall和 Friedman等[71-74]提出了等转化率动力学方法。等转化率法由于其简单性和低错误风险而被广泛使用。等转化率法可用于评估简单和复杂的动力学反应,不需要先明确反应机理,该动力学模型均基于以下表达式:

式中,α表示转化率,f(α)为反应机理函数。对于纤维素热解,α值通常在0.1-0.8选取,超出此范围的α值拟合效果则较差。
2.5.1 Kissinger法
该方法采用不同加热速率(β)进行多组实验,以ln(β/T2max)对1000/Tmax作图从而获得活化能的值,其中,Tmax是DTG曲线的峰值温度,表示最大分解温度。
其表达式为:

式中,g(α)表示反应机理常数的积分表达形式,在给定的转换值下被视为常数。通过线性拟合所得的斜率可计算出表观活化能,即-Ea/R[71]。
2.5.2 Flynn-Wall-Ozawa法(FWO)
该方法可简单表示为:
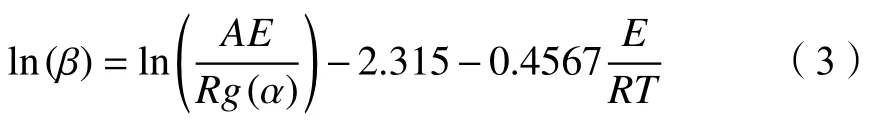
对于每种给定的α值,均可在不同升温速率β的热重曲线上找到该α值对应的T,而活化能Ea可以根据不同加热速率下ln(β)对1000/T作图拟合的斜率(-0.4567Ea/R)计算得出[72,73]。
2.5.3 Kissinger-Akahira-Sunose法(KAS)
KAS法又称广义Kissinger法,其表达式为:

该式中T值的获取方法与FWO法相同,对于给定的转化率α值,该方法以ln(β/T2)对1000/T的关系图计算表观活化能,斜率即为-Ea/r[71]。
2.5.4 Starink法
Starink法将FWO法和KAS法进一步修正为以下表达式:
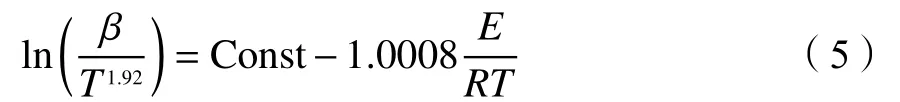
该方法的表观活化能通过ln(β/T1.92)对1000/T作图线性拟合计算即可得出[75]。
由于纤维素类型、实验条件、模型选取、转化率选取和线性区间选择的差异,不同研究获得的动力学参数有所不同。以Khan等利用以上四种模型所计算得出的纤维素热解表观活化能为例,如表1所示,除Kissinger法外,其他方法所得的表观活化能均与所选取的转化率α相关。通常转化率较低时,由于纤维素此时结晶度较高,结构单元之间结合紧密,因此,反应活化能较高。一般选取不同转化率α所得的表观活化能取平均值作为最终结论[76]。

表1 四种不同动力学方法计算出的纤维素热解表观活化能[76]Table 1 Ea of cellulose pyrolysis calculated by different kinetic methods[76]
3 纤维素的催化热解技术
近年来,围绕降低氧含量以改善生物油品质的关键问题已经开展了大量研究,其中,催化热解技术逐步引起研究人员的重视。基于催化剂结构特性的差异,可以选择性获得不同高值热解产物,其中,催化剂的脱氧特性能够显著改善生物油的物理和化学性质。因此,这方面研究主要倾向于设计高效催化材料用于提高特定高价值产物的选择性,如单环芳烃、酚类等,或者减少热解低值产物,如酸类、羰基化合物等。本章节将详细介绍纤维素催化热解方面的研究进展。
3.1 催化反应路径
如图9所示,纤维素催化热解涉及的主要化学反应包括脱氧、裂解、芳构化、酮化、羟醛缩合、加氢和重整等。通过催化剂结构性质调控和改变反应条件,可以选择性优化反应路径,提升热解产物品质。
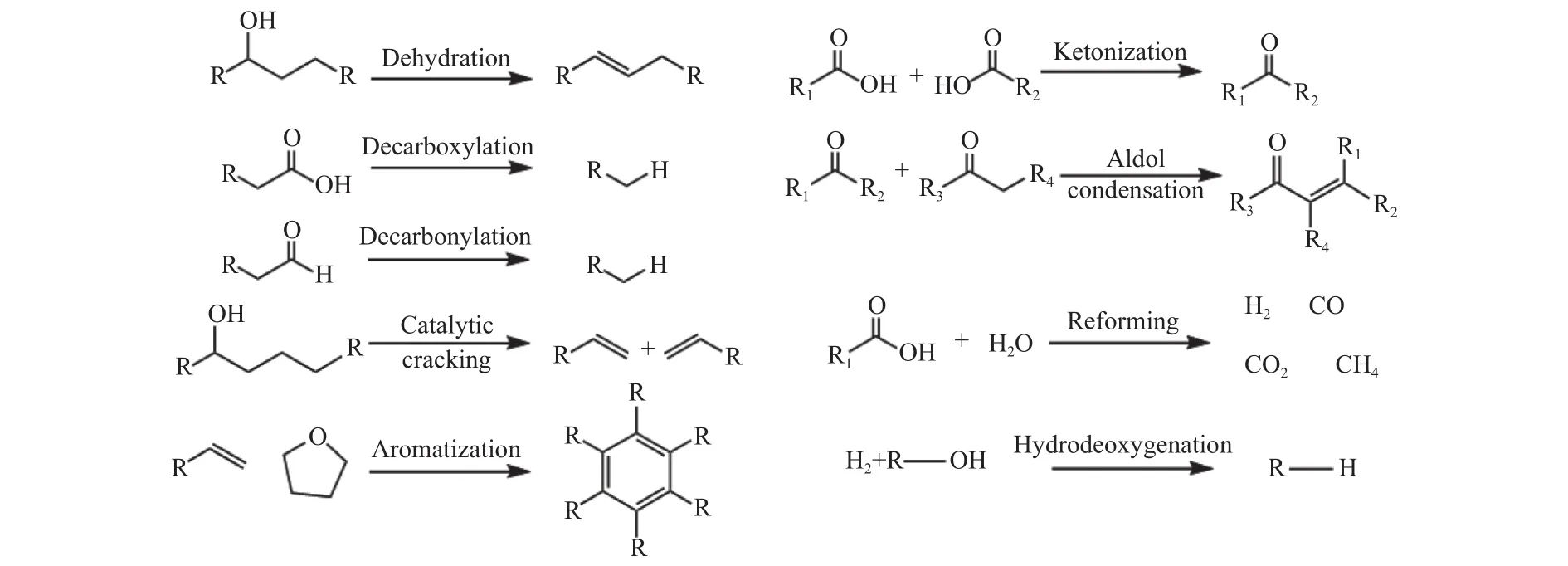
图9 纤维素催化热解反应路径示意图Figure 9 Reaction pathways for the catalytic pyrolysis of cellulose
催化脱氧是降低生物油氧含量、提升品质的有效途径。脱水、脱羧和脱羰是典型的脱氧反应,分别以H2O、CO和CO2的形式除去氧。其中,脱羰反应具有最高的脱氧效率,可以显著改善生物油的有效H/Ceff比。纤维素结构中糖单元脱水可以产生脱水糖和呋喃类化合物,如LG、LGO、DGP、FF 和 HMF等[77,78]。
催化裂解可以将有机大分子转化为小分子产物,特别是含氧化合物的催化裂解能够形成芳族化合物和烯烃。在催化裂解过程中,通常涉及碳碳键断裂、氢转移、异构化、芳族侧链断裂和脱氧等一系列反应。多元醇类化合物可通过反复脱水和氢转移反应产生烯烃,而烯烃与脱水中间体发生Diels-Alder缩合反应产生芳烃,低分子量化合物如酸和醇类的催化裂解会形成C1-C4烃类物质、H2O、CO和CO2[79]。
芳构化是将低分子量含氧化合物和烯烃转化为芳烃的另一种主要途径。在沸石分子筛作用下,酸、醇、醛、酯、醚和呋喃主要转化为芳烃[80]。芳构化过程可以用碳氢化合物池机理来解释:当中间含氧化合物扩散到沸石的孔道中时,它们经历一系列脱水、脱羧、脱羰和低聚反应,形成碳正离子烃池;在沸石和某些有机活性物质的协同作用下,烃池中间体可以进一步转化为烯烃、单环芳烃和多环芳烃中心,也可以用作助催化剂[81,82]。
酮化和羟醛缩合是将羧酸和羰基类热解组分转化为长链中间体的一种可能途径,可以进一步提质为汽油或柴油的取代燃料[83]。酮化反应是将两个羧酸分子转化为酮、CO2和H2O的反应,该反应可同时除去羧基并实现C-C偶联而不消耗外部氢,从而提高产物的热值和稳定性。其中,金属氧化物和沸石对羧酸酮化具有较高活性。酮化反应机理较复杂,乙烯酮、β-酮酸、羧酸盐和酰基碳离子被认为是关键的中间体。Brønsted酸和Lewis酸是该反应的有效催化剂,由于活性位点上的竞争性吸附,酸性位点的存在阻碍了醛醇缩合,对酮化反应更有利[84]。
重整和加氢脱氧也是改进生物油品质的有效途径。乙酸盐和羟基乙醛的蒸汽重整会产生部分H2。通过与氢气反应,生物油或其经过相分离后的有机相部分可以通过温和的加氢处理以降低氧含量,或者在更苛刻的加氢条件下直接转化为碳氢化合物。此外,还可以选择在原位催化快速热解过程中将催化剂和原料同时添加到热解反应器,在氢气气氛下直接将热解蒸汽转化为加氢脱氧产物。加氢脱氧可以保留产品中的大部分碳,同时在一定氢气压力下将产品中的氧组分转化为水[85,86]。
3.2 催化剂
3.2.1 无机盐离子催化剂
生物质本身含有无机矿物质,如K、Ca、Na和Mg等,这些无机物质通过改变纤维素分解的反应途径来影响快速热解产物的分布[87],具体如表2所示。碱金属和碱土金属(AAEMs)是生物质中最丰富的无机离子,它们对纤维素热解具有显著促进作用。钾盐的存在降低了纤维素热解的活化能和温度,有助于提高气体、水和焦炭的产率[88,89]。Saddawi等[90]研究发现钾盐促进了纤维素组分的解聚和碎片化,导致部分低分子量组分的选择性提高,如2(5H)-呋喃酮、糠醛等。此外,研究进一步证实钾盐能够显著抑制羰基化合物和醇类物质的形成,促进CO、CO2和H2O的形成,这主要是由于金属离子和含氧基团的相互作用促进了成环和裂解反应[90]。与KCl作用类似,NaCl亦能催化纤维素热解,形成大量低分子量产物。其中,NaCl不利于纤维素热解形成LG,这是因为钠离子的存在有利于竞争性脱水反应,抑制了纤维素端链的引发和脱除[91]。
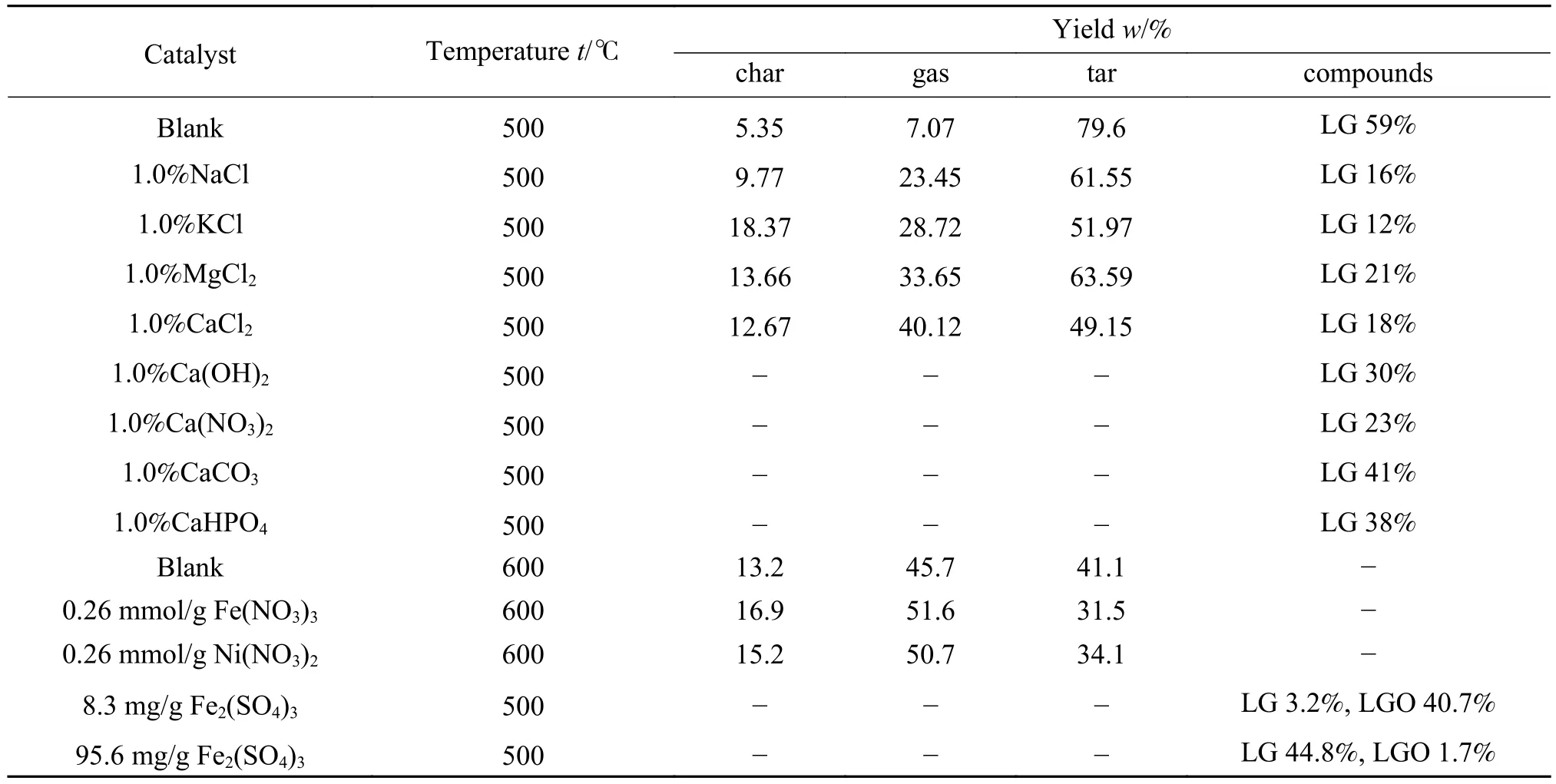
表2 不同类型的无机盐离子催化剂对纤维素热解产物分布的影响[87, 96]Table 2 Effect of different types of inorganic salt ion catalysts on the distribution of cellulose pyrolysis products[87, 96]
与碱金属离子(Na+和 K+)相比,碱土金属(Mg2+和Ca2+)氯化物Lewis酸性更强,能够促进糖类化合物脱水反应的进行。Liu等[92,93]系统研究了NaCl、KCl、MgCl2和 CaCl2对纤维素热解的催化作用,结果表明,Mg2+(MgCl2)和 Ca2+(CaCl2)对纤维素的热解影响明显大于Na+和K+。Patwardhan等[87]研究表明,AAEMs对纤维素热解形成LG的抑制作用如下:K+> Na+> Ca2+> Mg2+,而且 MgCl2和CaCl2的存在有利于生成呋喃环衍生物,如2-糠醛、HMF和LGO。
除碱金属和碱土金属外,研究人员发现一些过渡金属离子也具有一定的催化作用。ZnCl2是典型的路易斯酸,已被广泛用于纤维素催化热解研究。它可以催化C-C和C-O键断裂,有助于降解过程脱水、解聚和开环反应的发生。Carvalho等[94]研究发现ZnCl2的添加降低了纤维素最大降解速率,促进了焦炭和气体的形成,降低了液体产物收率。ZnCl2能同时催化热解过程中一次和二次反应的发生,通过戊糖基和葡萄糖基残基脱水,能够产生更多的糠醛和脱水糖。此外,ZnCl2可以和底物或产物中的水相互作用,部分水解产生HCl,进而促进催化热解反应发生。另一方面,水合形式的糖苷氧与锌的配位可以容易地切割糖苷键以形成单糖,并通过酸催化脱水反应进一步转化为脱水糖和呋喃化合物。Branca等[95]研究发现,ZnCl2亦可以作为脱水剂和交联剂,从而促进了碳化反应的发生。与Zn2+类似,Fe3+也具有催化纤维素转化为脱水糖的能力,通过吸收和离子交换将Fe3+引入纤维素后,LG和LGO的选择性分别最高达到44.8%和40.7%[96]。硝酸镍对结晶纤维素热解性能的影响与硝酸铁相近,铁和镍离子能够促进脱水和脱羧反应,抑制解聚过程,最终导致焦炭产率增加[37]。
除了金属阳离子影响之外,阴离子的存在也会影响纤维素热解行为。Mayer等[97]研究结果表明,浸渍硫酸盐的样品比浸渍有硝酸盐的样品具有更低的降解率和峰值温度。Patwardhan等[87]实验发现,在不同钙盐催化的纤维素热解过程中,LG 产率变化规律如下:Cl->≈ OH->≈。
3.2.2 金属氧化物催化剂
大多数金属氧化物及其混合物由于晶格缺陷(如电子空穴、点缺陷、位错和平面缺陷)而具有酸、碱或酸碱复合的性质。金属氧化物的表面酸碱性质可能有利于纤维素热解,选择性生成一些特定产物。金属氧化物的不同酸碱位点能够显著改变生物油组分分布,促进或抑制某些产物的形成[3]。表3给出了金属氧化物对纤维素热解产物分布的影响情况。

表3 不同类型的金属氧化物对纤维素热解产物分布的影响[1, 105]Table 3 Effects of various metal oxide catalysts on the product distribution for cellulose pyrolysis[1, 105]
Al2O3和SiO2是生物质催化热解中最常用的两种酸性金属氧化物。通常,酸性金属氧化物可促进有机分子的脱水、脱羰和裂化,降低生物油的氧含量并将大分子有机物转化为小分子组分。研究发现,酸性Al2O3的存在使生物油产率由58%降至40%左右,但是烃类和CO明显增加[1]。SiO2具有弱酸性和中等孔隙率,对去除含氧化合物(如酸、酮和醛)具有良好的催化活性,同时又能抑制焦炭和多环芳烃的形成。复合氧化物SiO2-Al2O3表现出与Al2O3相似的催化性能,但是产物中重质组分比例较高[1]。Li等[98]研究发现,水滑石结构的层状Ni-Sn双氧化物可选择性催化纤维素热解生成 1-羟基-3,6-二氧双环 [3.2.1]辛烷-2-酮(LAC)。金属氧化物的磺化可以增强催化剂酸性,并且能够选择性地催化特定化合物的形成。在纤维素热解中,/SnO2对5-甲基糠醛具有独特选择性,而/TiO2则有利于选择性生成糠醛。与纯ZrO2催化热解产物形成规律相反,/ZrO2催化剂上呋喃化合物的形成是建立在LG转化基础上,LG产率明显降低。磁性催化材料/TiO2-Fe3O4也被用于纤维素选择性热解制备LGO,最高收率可达15.43%,这主要归功于/TiO2-Fe3O4的强酸性位点[99-101]。
MgO和CaO是纤维素催化热解反应典型的碱性催化剂,主要通过酮化和醛醇缩合方式进行脱氧。碱性催化剂也可促进碎裂反应,从而形成低分子量产物。虽然热解过程MgO的添加降低了生物油产量,但是碳氢化合物的生成和含氧基团的去除却提高了生物油的热值[102]。Stefanidis等[83]研究了不同结构MgO的催化热解特性,结果表明,比表面积和孔隙率较大的MgO具有相对较好的催化活性。研究表明,CaO不仅具有显著的脱氧反应活性,还能促进烷基化环戊烯酮、烷基化酚和多环芳烃(PAH)的形成,极大地降低酸和脱水糖产率。当CaO和MgO混合使用时,热解生物油氧含量和酸度明显降低,但是总生物油产率较低,且最终热解产物中形成大量焦炭[103]。
与微米颗粒金属氧化物相比,纳米颗粒金属氧化物具有极高的比表面积,在纤维素催化热解过程中则表现出更优异的活性。TGA实验结果表明,与微米级NiO相比,纳米NiO能够进一步降低热解动力学分解温度、焦炭产率和反应活化能[104]。Fabbri等[105]利用各种纳米金属氧化物SiO2、Al2O3、MgO、TiSiO4和Al2O3-TiO2催化纤维素热解,研究其对手性脱水糖选择性的影响。结果表明,除纳米SiO2和MgO外,其余纳米催化剂均促进了脱水糖的形成。纳米氧化硅使脱水糖产率从20%减少到4%,而纳米氧化铝、硅酸钛和钛酸铝则将脱水糖总产率从20%分别提高到27%、28%和35%,其中LGO的产率从3.7%分别提高到7.7%、10%和19%[106]。Donar等[107]研究发现,SnO2纳米粒子在降低羰基含量、改善焦油稳定性和促进炭裂解反应方面具有较高的催化活性,产物中H2和小分子烃类物质(C1-C4)产率明显提高。Li等[108]采用ZrO2-SnO2纳米混合金属氧化物催化剂,将5-羟甲基糠醛(5-HMF)的产率提高到6.49%。
活性组分浸渍或制备二元金属氧化物等方法可以改变金属氧化物结构特性,包括孔道、孔隙率、比表面积、酸碱性等,从而调控它们的催化性能。Fe(Ⅲ)/CaO催化剂能够将重质酚类转化为侧链不含甲氧基和不饱和键的轻质酚类,其在提升呋喃、轻质脂肪烃和芳香烃收率的同时还降低了低值组分酸、醛和酮等的收率[109]。Fe/γ-Al2O3对H2具有良好的选择性,其占气态产物的30%-35%,这是由于Fe和Fe2+作为Fe/γ-Al2O3的活性中心促进了蒸汽重整和水煤气变换反应[110]。相比较单一金属氧化物,二元组分(MgAl、Mg2Al、Zn2Al)能够显著降低脱水糖产率,而金属氧化物物理混合后的催化效果与单一金属氧化物并无显著差异。不同金属原子比例和种类构成的二元金属氧化物催化热解纤维素的产物分布也各不相同,例如MgAl催化产生的呋喃类化合物比例最高,而Zn2Al更倾向于促进酚和脱水糖的生成,同时抑制小分子的产生[111]。由此可见,适当调变金属组成比例和种类是调节纤维素热解产物分布和优化产物选择性的关键。
3.2.3 沸石分子筛催化剂
沸石分子筛是纤维素催化热解研究最常用的一类固体酸催化剂[112]。沸石的主体结构由SiO4和[AlO4]-四面体构成,其中,[AlO4]-四面体的负电荷通过 H+补偿,形成 Brønsted 酸性位点[113]。沸石分子筛通常具有多维微孔结构,允许小分子扩散到沸石内部结构中的酸性位点,具有明显的形状选择性。各种分子筛依据孔径被分为三个大类:微孔(< 2 nm)、介孔(2-50 nm)和大孔(> 50 nm)[114]。其中,微孔分子筛具有良好的脱氧性能和对碳氢化合物的形状选择性,特别是质子化形态的HZSM-5,已被证实是最高效的热解制芳烃催化剂[115]。相比之下,由于活性位点高度暴露在基材上,介孔和大孔沸石不利于特定产物的生成。本节重点介绍HZSM-5催化纤维素热解研究进展。
当使用HZSM-5催化纤维素热解,热解蒸汽在催化剂的表面和孔道内经历了一系列反应,包括裂化、脱氧、脱羧、环化、芳构化、异构化、烷基化、歧化、低聚和聚合反应等[116],其可能的反应路径如图10所示。生物质热解过程各种含氧化合物在HZSM-5催化剂上的反应活性差异较大,醇类化合物在200 ℃左右迅速脱水形成相应的烯烃,并在较高温度(350 ℃)下进一步转化为高级烯烃和芳烃。相比之下,酮和酸的反应活性明显低于醇。丙酮脱水形成异丁烯,在高于350 ℃的温度下转化为C5+的烯烃、直链烷烃和芳烃,而乙酸通过以丙酮作为中间体,逐渐转化为烷烃和芳烃。乙醛和苯酚在HZSM-5上反应活性极低,催化剂床层中形成大量焦炭[117,118]。此外,具有各种官能团的含氧化合物脱氧过程在HZSM-5作用下反应机制也发生变化。其中,醇类和酚类主要以H2O的形式脱氧,醛类、甲酸盐类和碳水化合物主要以CO和H2O的形式脱氧,而羧酸则以CO2和H2O的形式脱氧[113]。

图10 HZSM-5催化纤维素热解反应途径示意图Figure 10 Reaction pathway for cellulose pyrolysis over HZSM-5
沸石分子筛的酸度和孔隙率显著影响生物质催化热解反应路径和产物分布。总酸度的增加有利于形成未取代的芳香族化合物(苯、萘、蒽等)和稠环芳香族化合物,同时抑制烷基取代芳烃(二甲苯、二甲基萘、甲基蒽等)的形成。因此,调整ZSM-5的酸度可以得到一系列优化的热解产物,以满足特定的需求。同时,还可以通过限制沸石的孔径调控反应中间体和产物。使用孔径过小的沸石催化纤维素热解主要产生CO、CO2和焦炭,沸石孔径过大则焦炭产率较高、芳烃和含氧化合物产率较低,只有孔径在5.2-5.9 Å的沸石,才有利于芳烃和烯烃的产生[119,120]。
如表4所示,用某些金属阳离子或氧化物对沸石分子筛进行改性可以调控热解产物分布及产率,而且通过调节酸性活性位点能够有效抑制焦炭的形成。Veses等[121]用 Mg、Ni、Cu、Ga和 Sn改性获得一系列M/ZSM-5(M为对应金属元素)。实验结果表明,改性后的ZSM-5显著改善了生物油的品质,降低了黏度和氧含量。他们还观察到酸性化合物和呋喃的产率降低,而碳氢化合物的产量进一步增加。Ni/ZSM-5和Sn/ZSM-5最具应用前景,它们既能够保证较低的焦炭形成速率,又可以有效增加碳氢化合物的产量。Mullen等[122]观察到Fe/ZSM-5显著改变了纤维素热解反应路径,以苯和萘的生成取代了对二甲苯和其他烷基苯的生成,同时抑制了苯酚的形成,显著降低焦炭产量。Ni/ZSM-5和Co/ZSM-5可促进芳烃和酚类化合物的形成。Co的加入显著提高了CO2产率,这表明掺杂Co有助于脱羧反应的发生。此外,Zn/ZSM-5催化剂亦在纤维素催化热解形成芳烃方面具有较好的活性,且能够较好地抑制热解过程焦炭的形成[122]。
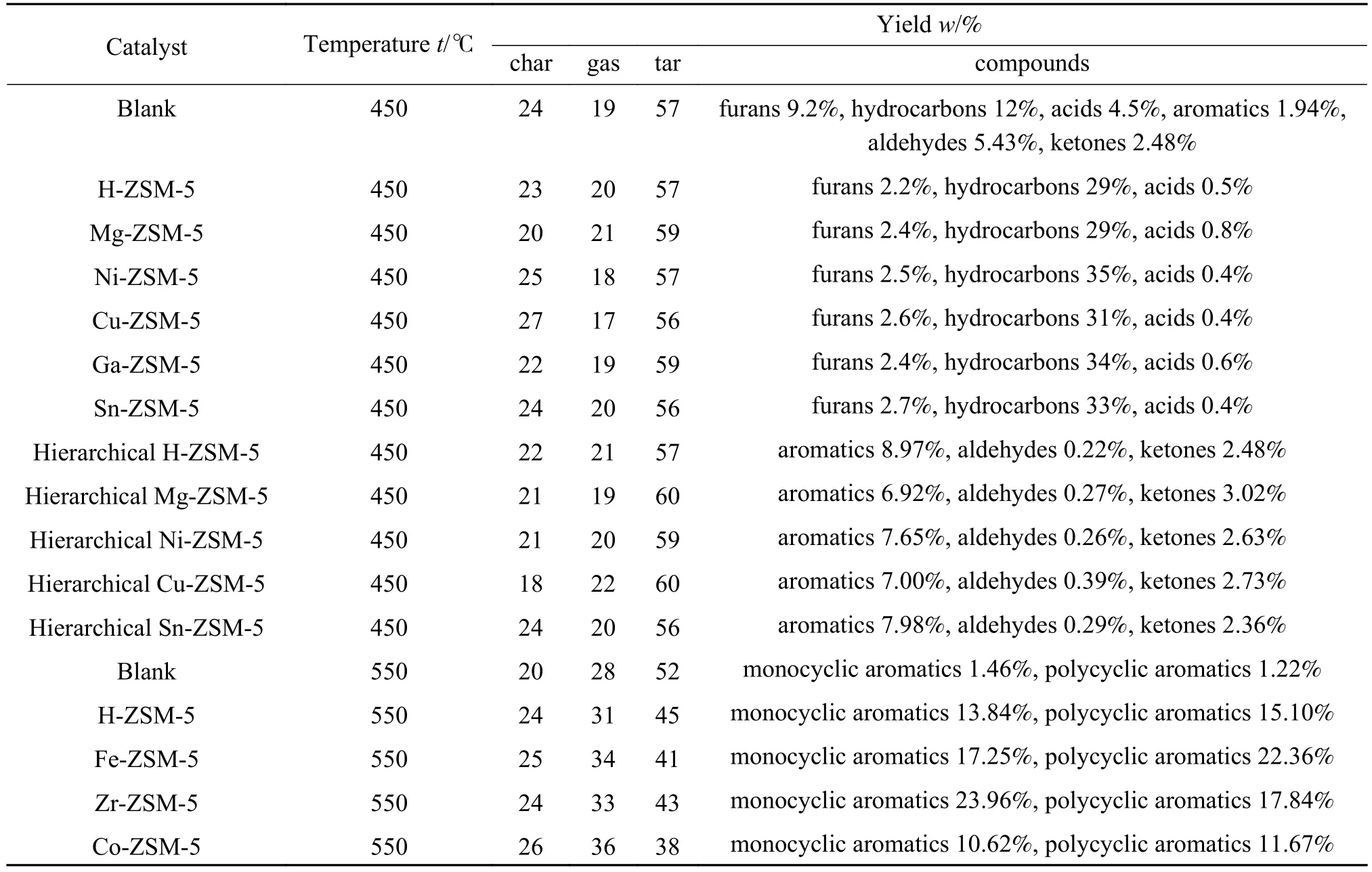
表4 不同类型的沸石分子筛催化剂对纤维素热解产物分布的影响[121, 122, 125]Table 4 Effect of different types of zeolite catalysts on the distribution of cellulose pyrolysis products[121, 122, 125]
ZSM-5沸石的微孔(孔径为5.2-5.9 Å)结构有利于选择性地催化合成芳烃。然而,狭窄的微孔不利于大分子含氧化合物进入孔道结构内,难以接触到活性位点,导致它们聚集在催化剂表面形成焦炭。此外,微孔系统还导致产物的扩散受到限制,可能会进一步促进焦炭形成和降低目标产物的实际产率。相比之下,大孔和中孔结构更有利于大分子的反应,但是作为HZSM-5催化剂的替代品却缺乏足够的尺寸选择性[3]。多级孔沸石具有典型的微孔-介孔-大孔结构,孔隙的分层结构促进了沸石晶体内物质的扩散,并使大分子顺利接触活性位点,因此,在热解蒸汽的脱氧过程中表现出明显的优势[123]。介孔表面Brønsted酸性位点的增加还会进一步促进芳族化合物的形成、降低含氧化合物含量。近年来,通过负载金属改性多级孔沸石用于热解反应方面已有大量研究报道。Kim等[124]开发了用于2-甲基-2-丁烯芳构化的多级孔Zn/ZSM-5催化剂,发现焦化反应被显著抑制。Neumann等[125]观察到铈掺杂多级孔HZSM-5催化剂可显著抑制葡萄糖热解过程焦炭的形成。然而,铈掺入后引起催化剂酸量降低,产物中含氧化学品明显增加。Veses等[126]发现Sn、Cu、Ni和 Mg改性的多级孔ZSM-5沸石可以促进脱氧反应,并且 Mg2+的加入产生了路易斯酸位点,促进了酸与醛发生酮化反应产生酮类产物。
对于沸石分子筛上纤维素催化热解反应,焦炭和无机物在催化剂表面沉积导致其失活是目前最具挑战性的问题。这种复合失活过程受许多因素的影响,包括原料组成、反应条件和沸石分子筛的性质等。HZSM-5外部的强酸性位点是催化剂上大分子含氧化合物在表面聚合形成焦炭的主要原因,因此,通过降低外部强酸性位点、保留内部酸性位点的表面钝化技术已被证明是降低焦炭产率的有效手段[127]。化学液相沉积(CLD)是一种常见的催化剂外表面改性方法,可以调变表面结构,减少催化剂表面酸性位点[128]。此外,在沸石分子筛表面进行预结焦是抑制焦炭产率的另一种有效方法。Zhang等[129]发现,使用过的HZSM-5沸石能够最大幅度降低焦炭的产率,并且有利于形成具有比新制沸石更高的烃含量和更低氧含量的目标产物。Wang等[130]通过甲醇-烯烃反应预焦化改性HZSM-5沸石,降低了其外部强酸性位点和比表面积,预焦化百分比提高至5.4%。相应地,随着预焦化百分比的增加,焦炭产率逐渐降低,而烃类物质产率则先增加后减少,其最大值为53.47%。在催化热解过程中,一部分无机矿物质可能随着热解蒸汽蒸发并沉积在催化剂表面,使催化性能劣化或甚至导致催化剂失活。Wang等[131]研究了碱金属和碱土金属对沸石催化剂上生物质异位催化热解的影响,发现痕量AAEM也会显著降低烃产率,且催化作用依次为 K+> Na+> Ca2+> Mg2+。同时,灰分的加入降低了有机物和焦炭的产率,增加了水和不可冷凝气体的产率。提前浸出生物质原料中的无机盐离子,或者在催化剂再生前先除去焦炭,则可以在一定程度上克服快速催化热解中原材料灰分带来的缺点。除了上述已有研究,还需要进一步探讨无机盐在生物质催化热解中的作用,这对于揭示热解蒸汽、无机盐和催化剂三者之间的相互作用及其对催化剂寿命的影响至关重要。
3.3 原位和非原位催化热解
除了催化剂种类之外,原料和催化剂的接触方式亦会影响纤维素热解过程产物分布。根据两者接触方式的不同,快速催化热解分为原位催化热解和非原位催化热解(串联催化热解)两种形式。将催化剂与热解反应器中的原料直接混合后进行热解称为原位催化热解,本质是催化剂和纤维素混合,改变纤维素热解反应历程。而将热解所得蒸汽输送到下游的单独催化剂床再进行催化反应称为非原位催化热解,本质是利用催化剂影响挥发性热解产物发生的二次反应[132]。
原位催化热解的操作简单,反应器的结构无需特别调整,有利于热解碎片释放后立刻进行裂解,同时最大限度地减少热解初级产物的二次裂解,能够在起到降低热解起始温度的同时获得比非原位催化热解更高的芳烃产率[133]。其缺点是催化剂失活较快,大量焦炭和无机物附着在用完的催化剂上导致催化剂难以再生,而且原位催化热解时纤维素热解和催化转化在同一温度进行,这在一定程度上限制了这两步反应的温度参数优化。
非原位催化热解的催化剂寿命较长,不会由于无机物附着导致催化位点的永久失活,而且产生的焦炭很少,易于进行催化剂的再生操作。非原位催化热解的每个步骤都可以分别在最佳条件下进行,这有利于生物油品质的提升[132]。然而它的缺点主要在于反应器的设计方面,非原位催化热解需要一个单独进行加热的催化床,还需要保持催化床和反应物之间的温度稳定,因而设计成本比原位催化热解要高。在需要将无机盐等催化剂通过吸收或离子交换浸渍到原料中时,原位催化热解也优于非原位催化热解[132]。
原位和非原位催化热解都可以显著提高纤维素热解生物油品质。Li等[134]对用于生物燃料生产的原位和非原位催化裂解进行了技术经济和不确定性分析。其经济评估表明,原位催化裂解的最低燃料销售价格低于非原位催化热解,而非原位催化的技术经济不确定性低于原位催化。并非所有类型的催化热解都可以自由选择原位和非原位催化两种形式,其选取还要取决于催化剂的性状等因素。例如,使用无机盐离子催化剂进行催化热解需要将热解原料置于催化剂溶液中浸渍处理,此种情况则只能使用原位催化热解的形式。反应器的配置在催化热解技术发展中起重要作用,非原位和原位催化热解均有各自的优点和缺点。因此,在为特定研究选择反应器配置时,应全面考虑原料类型、所需产物和操作条件等因素。
4 结 论
本综述详细介绍了纤维素热解的原理和技术方法,系统对比了纤维素热解的各种分析方法和催化剂的优势与不足,总结了以优化产物分布并提高热解生物油品质为研究目的的发展方向。过往研究表明,热解生物油选择性分离困难、催化剂失活快并且难以再生是制约纤维素热解技术应用的重要原因,而目前单纯依靠反应体系和常规催化剂等技术方法依然难以改变生物油组分的复杂性,并且在催化剂的使用寿命成本等方面仍然存在技术瓶颈。因此,纤维素热解的未来研究方向还需要更加注重于理论的创新,加深对热解时纤维素结构转变与产物选择性之间关系的认识,要围绕新的热解理论设计催化反应体系,在获得合适产物分布的同时还要侧重于目标产物的有效分离提纯,通过多方面的协同配合才能提高热解技术的经济价值,最终实现热解技术高效广泛的实际应用。纤维素热解技术研究的发展将会成为由纤维素制取各种高值化学品和可再生能源的关键技术,为化石能源的替代提供了一条重要的可行途径,有利于未来资源的可持续发展。
