3-苄基-5-(1-(2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基)亚乙基)-2-氨基咪唑啉-4-酮类化合物的合成及杀菌活性
2021-12-23李益豪许磊川苏彦豪王明安
李益豪, 赵 宇, 许磊川, 苏彦豪, 张 倩, 王明安
(中国农业大学 应用化学系 农药创新研究中心,北京 100193)
螺环丁烯内酯类化合物广泛存在于自然界中,如spirofragilide[1]、lambertellol A[2]、crossalactone D[3]和pyrenolide D[3]等,这些天然产物具有杀虫[4]、杀菌[5]、抑制孢子萌发[6]、抗炎[7]和抗肿瘤[8]等多种优良的生物活性,已商品化的螺环丁烯内酯类农药品种包括螺螨酯 (spirodiclofen)、螺甲螨酯(spiromesifen)、螺虫乙酯 (spirotetramat) 和甲氧哌啶乙酯 (spiropidion)[9-12],如图式1 所示,可见螺环丁烯内酯是一个值得深入研究的优异药效团。
咪唑啉酮类化合物具有优良的杀菌活性[13-17],如已经商品化的农用杀菌剂咪唑菌酮 (fenamidone)是一种线粒体呼吸抑制剂,通过抑制病原菌线粒体呼吸作用进而抑制能量的生成而达到杀菌作用,其作用机制独特,杀菌谱广,对葡萄及蔬菜霜霉病、马铃薯及番茄晚疫病等卵菌病害菌株有效[18]。在对咪唑菌酮的结构改造过程中发现,当咪唑啉酮3-位被苄基取代时,也表现出优异的杀菌活性[19-21]。本课题组前期采用多样性导向合成的方法,以活性亚结构拼接的原理将螺环丁烯内酯和咪唑啉酮等多种药效团进行组合,设计合成了包括5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-氨基咪唑啉-4-酮在内的多类化合物,生物活性评价结果显示,部分化合物显示出优异的杀菌和杀虫活性[22-30],但是发现这类2-氨基咪唑啉酮类化合物在溶液中存在多种互变异构体,不便于表征其化学结构和探讨其构效关系[22-24]。为了发现更高杀菌活性的该类化合物并减少互变异构的可能,本文在前期工作的基础上,参考3-苄基咪唑菌酮的结构,期望在咪唑啉酮环的3-位引入苄基,设计合成一系列新型3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-氨基咪唑啉-4-酮类化合物 (设计策略见图式2),对其进行结构表征并评价其杀菌活性。合成路线见图式3。
1 实验部分
1.1 仪器、试剂和药剂
Bruker DPX 300 和Ascend 500 MHz 核磁共振仪 (以CDCl3为溶剂,TMS 为内标);WRX-4 显微熔点仪 (上海易测仪器设备有限公司,温度未校正);岛津高效液相色谱LC-20AT, Luna C18反相色谱柱 (250 mm × 4.6 mm, 5 μm, Phenomenex,Torrance, CA, USA),流动相为甲醇/水 (V甲醇:V水=60 : 40),流速1.0 mL/min, 紫外检测波长230 nm。SHB-III 循环式真空泵 (郑州恒岩仪器有限公司);EYELA N-1100 旋转蒸发仪 (北京天林恒泰科技有限公司);DF-101S 集热式恒温磁力搅拌器(郑州长城科工贸有限公司)。
柱层析硅胶为青岛海洋化工有限公司生产(200~300 目);试剂均为国产或进口分析纯,无水溶剂用常规方法干燥处理;98%多菌灵 (carbendazim) 原药,上海阿拉丁生化科技股份有限公司;98%咪唑菌酮 (fenamidone) 原药,拜耳作物科学公司。
1.2 目标化合物的合成
1.2.1 3-乙酰基-4-甲基-1-氧螺[4,5]癸-3-烯-2-酮(2) 的合成 参照前文方法合成,得到白色固体,收率53%,m. p. 98~99 ℃,其1H NMR 和熔点数据与前文[23]一致。
1.2.2 3-苄基-2-硫代乙内酰脲 (3) 的合成 参照文献方法[31]合成,得到黄色固体,收率91%,m. p.191~193 ℃,其1H NMR 和熔点数据与文献[31]一致。
1.2.3 化合物4 和5 的合成 在100 mL 圆底瓶中,依次加入1.50 g (7.2 mmol) 化合物2、1.48 g(7.2 mmol) 化合物3、40 mL 甲苯和1 mL 三乙胺,缓慢加入0.66 g (10.8 mmol) 乙醇胺,110 ℃回流搅拌反应6 h。薄层层析 (展开剂为V正己烷:V乙酸乙酯=5 : 1) 监测至原料点消失。将反应液冷却至室温后减压旋蒸除去甲苯,粗产物经硅胶柱层析 (V正己烷:V乙酸乙酯= 8 : 1) 分离,得到202 mg 淡黄色固体化合物4 和1.53 g 淡黄色固体化合物5。
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-硫代-2,4-咪唑二酮 (4):淡黄色固体,收率7%,m.p. 157~159 ℃.1H NMR (300 MHz, CDCl3),δ:8.75 (s, 1H), 7.50 (d,J= 5.0 Hz, 2H), 7.35-7.27 (m, 3H), 5.05(s, 2H), 2.40 (s, 3H), 2.00 (s, 3H), 1.80-1.25 (m, 10H);13C NMR (75 MHz, CDCl3),δ: 176.57, 170.73, 170.27, 162.69,135.68, 129.10, 128.66, 128.08, 126.82, 125.39, 118.75, 89.15,44.75, 33.66, 24.50, 21.93, 16.81, 13.29. HR-ESI-MS:C22H25N2O3S [M+H]+,计算值397.158 0,测试值397.158 4.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-硫代-2,4-咪唑二酮 (5):淡黄色固体,收率53%,m.p. 179~181 ℃.1H NMR (300 MHz, CDCl3),δ:9.66 (s, 1H), 7.39 (d,J= 5.0 Hz, 2H), 7.32-7.20 (m, 3H), 4.99(s, 2H), 2.10 (s, 3H), 1.87 (s, 3H), 1.83-1.20 (m, 10H).13C NMR (75 MHz, CDCl3),δ: 176.95, 170.78, 169.28, 160.55,135.55, 128.65, 128.58, 127.93, 127.71, 123.37, 118.13, 88.60,44.49, 33.61, 33.23, 24.75, 22.02, 18.89, 12.91. HR-ESI-MS:C22H25N2O3S [M+H]+,计算值397.158 0,测试值397.158 6.
1.2.4 化合物6 和7 的合成及构型转化 在50 mL圆底瓶中,加入200 mg (0.5 mmol) 化合物4、104 mg(0.75 mmol) 碳酸钾和20 mL 乙腈,搅拌下缓慢滴加107 mg (0.75 mmol) 碘甲烷,室温反应6 h。薄层层析 (展开剂为V石油醚:V乙酸乙酯= 4 : 1) 监测至原料点消失。过滤除去碳酸钾,旋转蒸发除去乙腈,用乙酸乙酯重结晶得到188 mg 黄色固体化合物6。以5 为原料,采用相同的方法合成化合物7。
(Z)-3-苄基-2-甲硫基-5-(4-甲基-2-氧代-1-氧杂螺[4,5]-癸-3-烯-3-亚乙基)-咪唑啉-4-酮 (6):黄色固体, 188 mg,收率91%,m.p. 171~173 ℃.1H NMR (300 MHz, CDCl3),δ:7.35-7.27 (m, 5H), 4.71 (s, 2H), 2.57 (s, 3H), 2.42 (s, 3H), 1.87(s, 3H), 1.86-1.25 (m, 10H);13C NMR (75 MHz, CDCl3),δ:170.72, 168.41, 168.03, 161.24, 138.15, 135.86, 133.22,128.79, 128.04, 126.06, 87.84, 44.20, 33.69, 24.79, 22.09,16.18, 13.76, 12.85. HR-ESI-MS: C23H27N2O3S [M+H]+,计算值411.173 7,测试值411.173 8.
(E)-3-苄基-2-甲硫基-5-(4-甲基-2-氧代-1-氧杂螺[4,5]-癸-3-烯-3-亚乙基)-咪唑啉-4-酮 (7):黄色固体, 1 500 mg,收率92%,m.p. 193~194 ℃.1H NMR (300 MHz, CDCl3),δ:7.34-7.22 (m, 5H), 4.69 (s, 2H), 2.61 (s, 3H), 2.40 (s, 3H), 1.91(s, 3H), 1.85-1.20 (m, 10H);13C NMR (75 MHz, CDCl3),δ:170.82, 168.23, 166.09, 162.67, 139.60, 135.82, 131.41,128.75, 127.94, 127.70, 124.50, 88.14, 44.03, 33.76, 33.36,24.88, 22.12, 19.13, 12.91, 12.66. HR-ESI-MS: C23H27N2O3S[M+H]+,计算值411.173 7,测试值411.174 0.
化合物6 和7 的构型转化:将20 mgZ-构型纯度为95%的化合物6 加入到含有少量醋酸的甲苯溶液中,搅拌回流,每隔2 h 取少量反应液旋转蒸发除去溶剂后,以色谱纯甲醇溶解样品,用高效液相色谱 (HPLC) 仪进行Z/E-构型比例变化的分析检测。采用相同的方法对化合物7 的构型转化进行分析。
1.2.5 目标化合物8a~8f 和9a~9h 的合成 分别以化合物8a 和9a 的合成为例:在50 mL 圆底瓶中依次加入300 mg (0.73 mmol) 化合物7、102 mg(1.1 mmol) 苯胺、1.5 mL 冰醋酸和13.5 mL 甲苯,110 ℃回流搅拌反应8~10 h,薄层层析 (展开剂为V乙酸乙酯:V石油醚= 1 : 3) 监测至原料点消失。冷却至室温后减压旋转蒸发除去溶剂,粗产物经硅胶柱层析 (V乙酸乙酯:V石油醚=1 : 8~1 : 3) 梯度洗脱,得到100 mg 米黄色固体化合物8a,收率30%和127 mg 淡黄色固体化合物9a,收率38%。以200 mg 化合物6 为原料采用相同方法与苯胺进行反应,薄层层析 (展开剂为V乙酸乙酯:V石油醚=1 : 3) 监测所得产物与化合物8a 和9a 的极性相同,经硅胶柱层析得到80 mg 米黄色固体化合物8a 和75 mg 淡黄色固体化合物9a,核磁共振氢谱进一步证明为化合物8a 和9a。采用相同方法以7 为原料合成化合物8b~8f 和9b~9h。
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-苯基氨基-咪唑啉-4-酮 (8a):黄色固体,收率30%,m.p. 193~194 ℃.1H NMR (300 MHz, CDCl3),δ:7.60-6.90 (m, 10H), 4.93, 4.92 (s, 2H), 2.60, 2.38 (s, 3H), 1.90,1.87 (s, 3H), 1.75-1.20 (m, 10H). HR-ESI-MS: C28H30N3O3[M+H]+,计算值456.228 2,测试值456.228 5.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-苯基氨基-咪唑啉-4-酮 (9a):淡黄色固体,收率38%,m.p. 190~191 ℃.1H NMR (300 MHz, CDCl3),δ:7.61-6.96 (m, 10H), 6.46 (s, 1H), 4.96-4.70 (m, 2H), 2.22 (s,1H), 1.93-1.20 (m, 15H). HR-ESI-MS: C28H30N3O3[M+H]+,计算值456.228 2,测试值456.228 0.
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(3-氟苯基) 氨基-咪唑啉-4-酮 (8b):淡黄色固体,收率23%,m.p. 182~184 ℃.1H NMR (300 MHz,CDCl3),δ: 7.74-7.01 (m, 7H), 6.84-6.51 (m, 3H), 4.90 (s, 2H),2.58, 2.37 (s, 3H), 1.93, 1.87 (s, 3H), 1.85-1.22 (m, 10H). HRESI-MS: C28H29FN3O3[M+H]+,计算值474.218 7,测试值474.218 3.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(3-氟苯基) 氨基-咪唑啉-4-酮 (9b):白色固体,收率44%,m.p. 221~223 ℃.1H NMR (300 MHz,CDCl3),δ: 7.75-6.60 (m, 9H), 6.46 (s, 1H), 4.98-4.67 (m, 2H),2.16 (s, 1H), 1.90-1.21 (m, 15H). HR-ESI-MS: C28H29FN3O3[M+H]+,计算值474.218 7,测试值474.218 6.
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-甲氧基苯基) 氨基-咪唑啉-4-酮 (8c):红色固体,收率10%,m.p. 169~171 ℃.1H NMR (300 MHz,CDCl3),δ: 7.59-7.28 (m, 6H), 7.04-6.57 (m, 4H), 4.90 (s, 2H),3.77, 3.74 (s, 3H), 2.57, 2.36 (s, 3H), 1.90 (s, 3H), 1.87-1.19 (m,10H). HR-ESI-MS: C29H32N3O4[M+H]+,计算值486.238 7,测试值486.238 2.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-甲氧基苯基) 氨基-咪唑啉-4-酮 (9c):淡黄色固体,收率58%,m.p. 185~187 ℃.1H NMR (300 MHz, CDCl3),δ: 7.52-6.52 (m, 9H), 6.46 (s, 1H), 4.90-4.74(m, 2H), 3.81 (s, 3H), 2.28 (s, 1H), 2.00-1.20 (m, 15H). HRESI-MS: C29H32N3O4[M+H]+,计算值486.238 7,测试值486.238 5.
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-叔丁基苯基) 氨基-咪唑啉-4-酮 (8d):黄色固体,收率29%,m.p. 177~178 ℃.1H NMR (300 MHz,CDCl3),δ: 7.59-6.86 (m, 9H), 6.60(s, 1H), 4.91, 4.88 (s, 2H),2.58, 2.36 (s, 3H), 1.88, 1.86 (s, 3H), 1.82-1.30 (m, 10H), 1.29, 1.25(s, 9H). HR-ESI-MS: C32H38N3O3[M+H]+,计算值512.290 8,测试值512.290 4.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-叔丁基苯基) 氨基-咪唑啉-4-酮 (9d):白色固体,收率45%,m.p. 191~192 ℃.1H NMR (300 MHz,CDCl3),δ: 7.49-7.27 (m, 8H), 6.92 (d,J= 8.5 Hz, 1H), 6.51 (s,1H), 4.93-4.72 (m, 2H), 2.27 (s, 1H), 1.95-1.45 (m, 15H), 1.33, 1.27(s, 9H). HR-ESI-MS: C32H38N3O3[M+H]+,计算值512.290 8,测试值512.290 3.
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-溴苯基) 氨基-咪唑啉-4-酮 (8e):红色固体,收率24%,m.p. 198~199 ℃.1H NMR (300 MHz,CDCl3),δ: 7.54-7.15 (m, 7H), 6.85 (d,J= 8.6 Hz, 2H), 4.90 (s,2H), 2.59, 2.37 (s, 3H), 1.94, 1.82 (s, 3H), 1.81-1.24 (m, 10H).HR-ESI-MS: C28H29BrN3O3[M+H]+,计算值534.138 7,测试值534.139 6.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-溴苯基) 氨基-咪唑啉-4-酮 (9e):白色固体,收率48%,m.p. 209~211 ℃.1H NMR (300 MHz,CDCl3),δ: 7.58-7.28 (m, 8H), 6.86 (d,J= 8.5 Hz, 1H), 6.43 (s,1H), 4.99-4.65 (m, 2H), 2.16 (s, 1H), 1.95-1.21 (m, 15H). HRESI-MS: C28H29BrN3O3[M+H]+,计算值534.138 7,测试值534.139 3.
(Z)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-甲基苯基) 氨基-咪唑啉-4-酮 (8f):淡黄色固体,收率29%,m.p. 192~194 ℃.1H NMR (300 MHz,CDCl3),δ: 7.56-6.84 (m, 9H), 4.91, 4.88 (s, 3H), 2.58, 2.36 (s,3H), 2.30, 2.25 (s, 3H), 1.89, 1.86 (s, 3H), 1.80-1.20 (m, 10H).HR-ESI-MS: C29H32N3O3[M+H]+,计算值470.243 8,测试值470.244 1.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-甲基苯基) 氨基-咪唑啉-4-酮 (9f):白色固体,收率46%,m.p. 168~169 ℃.1H NMR (300 MHz,CDCl3),δ: 7.50-7.02 (m, 8H), 6.87 (d,J= 8.1 Hz, 1H), 6.44 (s,1H), 4.94-4.70 (m, 2H), 2.34, 2.27 (s, 3H), 2.25 (s, 1H), 1.90,1.85 (s, 3H), 1.82-1.25 (m, 12H). HR-ESI-MS: C29H32N3O3[M+H]+,计算值470.243 8,测试值470.243 5.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-三氟甲基苯基) 氨基-咪唑啉-4-酮 (9g):红色固体,收率82%,m.p. 218~219 ℃.1H NMR (300 MHz, CDCl3),δ: 7.75-7.04 (m, 9H), 6.44 (s, 1H), 5.02-4.67(m, 2H), 2.16 (s, 1H), 1.90-1.20 (m, 15H). HR-ESI-MS:C29H29F3N3O3[M+H]+,计算值524.215 6,测试值524.216 2.
(E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-氟苯基) 氨基-咪唑啉-4-酮 (9h):红色固体,收率80%,m.p. 188~190 ℃.1H NMR (300 MHz,CDCl3),δ: 7.60-6.87 (m, 9H), 6.39 (s, 1H), 4.99-4.60 (m, 2H),2.17 (s, 1H), 1.89-1.25 (m, 15H). HR-ESI-MS: C28H29FN3O3[M+H]+,计算值474.218 7,测试值474.218 2.
1.3 量子化学计算
在Gaussian View 程序中建立3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-苯基氨基咪唑啉-4-酮 (8a 和9a) 分子模型,将其保存为高斯输入文件,将该文件直接上传至Gaussian 09W 程序中,计算该化合物各互变异构体的优化结构。采用量子化学的密度泛函理论 (DFT) B3LYP方法,在B3LYP/6-31G (d, p) 水平上进行分子结构优化,得到理论计算的全优化构型,并对优化结果计算频率,振动分析表明所得优化构型均对应势能面上能量最小点 (即无虚振动频率),为稳定构型,计算收敛精度采取程序设定的缺省值。全部计算利用Gaussian 09W 软件在微机上进行。
1.4 杀菌活性测定
1.4.1 离体杀菌活性测试 供试菌株:棉花立枯丝核菌Rhizoctonia solani、番茄灰霉病菌Botrytis cinerea、辣椒疫霉病菌Phytophthora capsici、油菜菌核病菌Sclerotinia sclerotiorum和小麦赤霉病菌Fusarium graminearum,均由中国农业大学植物保护学院植物病理系分离并保存,活性测试时需要进一步活化。
采用菌丝生长速率法[32]测定。以DMSO 为溶剂,先将供试化合物配制成质量浓度为5 000 mg/L的药液,再以马铃薯葡萄糖琼脂液体 (PDA) 培养基稀释,制备成50 mg/L 的含药PDA 平板供试。将菌饼接种于含药PDA 培养基中央,置于24 ℃培养箱中黑暗培养2~3 d,以十字交叉法测量各处理的菌落直径。每个样品平行测定3 次,取平均值。以多菌灵和咪唑菌酮为对照药剂。以空白对照菌落增长直径和药剂处理的菌落增长直径的差值与空白对照菌落增长直径的比值来计算各药剂处理对各种病原菌的菌丝生长抑制率。EC50测定时,根据抑制率不同在100~0.1 mg/L 之间设置5 个浓度梯度。
1.4.2 活体杀菌活性测试 供试菌株:黄瓜霜霉病菌Pseudoperonospora cubensis、小麦白粉病菌Erysiphe graminis和玉米锈病菌Puccinia sorghiSchw.,由沈阳化工研究院分离并保存。
采用活体盆栽法[33-34]测定。将待测样品先用少量溶剂DMF 溶解,再用含有0.1%吐温-80 的水稀释,配制成质量浓度为400 mg/L 的药液。在小型喷雾塔上,将药液喷施于病害寄主植物上 (寄主植物为在温室内培养的标准盆栽苗,新泰密刺黄瓜一叶一心期,辽春10 号小麦二叶期,金黄糯2 号玉米2~3 叶期),24 h 后接种供试菌株,置于人工气候室培养,待叶片被侵染后移入温室继续培养,大约7 d 后进行防治效果评估。每个样品平行测定3 次,取平均值,以氟醚菌胺和氰霜唑为对照药剂。活体活性测试由沈阳中化化工有限公司新农药创制国家重点实验室参照前文方法[33-34]协助完成。
2 结果与讨论
2.1 化合物的合成、双键构型及产物构型的转化
参照文献方法,以环己酮和乙烯基乙醚为原料合成α-羟基酮 (1),在碱性条件下1 再与双乙烯酮反应合成2;采用异硫氰酸苄酯和甘氨酸甲酯盐酸盐为原料制备3;化合物2 和3 通过缩合反应分离得到具有Z-和E-构型的化合物4 和5。实验室前期所合成的此类化合物均为E-构型化合物[23-24],本研究中,化合物5 的两个特征甲基化学位移 (δ)在2.10 和1.87,与前文[15]中具有E-构型的类似中间体δ2.02 和1.85 基本一致,可以确定化合物5 也具有E-构型;而化合物4 的两个特征甲基δ在低场的2.40 和2.00,由此确定化合物4 具有Z-构型。本文首次分离得到Z-构型的该类化合物。分别将4 和5 在碱性条件下用碘甲烷进行甲基化得到化合物6 和7,该反应不涉及双键构型的变化,因此6 和7 也分别具有Z-和E-构型。它们在冰醋酸催化下与各种取代苯胺进行衍生化反应,得到目标化合物8a~8f 和9a~9h。值得注意的是,化合物7 分别与对氟甲基苯胺和对氟苯胺反应后只分离到化合物9g 和9h。
有趣的是,在缩合反应结束进行硅胶柱层析分离时,仅分离得到少量的Z-构型产物4,说明该反应仍然以相对更加稳定的E-构型产物5 为主要产物。在碱性条件下,在用碘甲烷分别对4 和5 进行甲基化制备化合物6 和7 时,产物双键构型保持不变;而在以化合物6 和7 为原料进行衍生化反应时,意外发现均会得到不同比例的Z/E-构型两种产物,但仍然以E-构型产物9 为主要产物。由于衍生化反应以醋酸作为催化剂,推测E-或Z-构型化合物在酸性条件下可以发生部分的构型转化,转化为相应的Z-或E-构型化合物,达到动力学平衡状态。为了证明这一推测的合理性,用HPLC 方法对中间体6 和7 在酸性条件下Z/E-构型比例的变化进行了分析检测,结果如图1 所示。HPLC 的分析结果证明,在酸性条件下Z-构型产物6 会转化为E-构型产物7,E-构型产物7 也会转化为Z-构型产物6,但大约6 h 后在反应平衡时总是以能量上更稳定的E-构型产物7 为主要存在形式,所以最终生成E-构型产物9 为主要产物。
经过文献检索发现存在类似的情况[35],推断构型转化原因可能是E-构型化合物6 的咪唑啉酮1-位氮原子在酸性条件下发生质子化,导致咪唑啉酮开环,发生构型转化后,再关环成E-构型化合物7,见图式4 所示。这种构型转化的现象在没有3-苄基的同类反应中并不明显。
2.2 化合物的氢谱解析和互变异构分析
目标化合物的结构经1H NMR 和高分辨质谱进行了表征。通过咪唑啉酮3-位苄基的引入,希望减少互变异构的可能性,实际上通过Z-构型6 或E-构型7 衍生化所得到的产物8a~8f 和9a~9h,可以通过柱层析进行有效的分离,但是8a~8f 和9a~9h 在溶液中仍然存在互变异构体。δ在4.0~5.0 之间的亚甲基质子和δ在2.0~3.0 范围的一个甲基质子是最特征的1H NMR 谱线,易于识别,可用于互变异构体的相对量分析,而其余质子在低场的芳环区以及高场的饱和碳上质子区相互重叠,难以区分。分离得到的Z-构型产物8 为两种互变异构体的混合物,n(8-1) :n(8-2) 在1.0 : 2.5~1.0 : 4.0 之间,两种互变异构体苄基的亚甲基部分都为单峰并基本重叠在一起,说明这两个异构体的两个亚甲基质子为等价质子 (图2)。在图式5 所示的8a-1 和8a-2 两种结构中,3-苄基所在的C—N 单键可以自由旋转,使两个亚甲基质子成为等价质子呈现单峰与实际谱图一致,因此化合物8a 在溶液中主要为8a-1 和8a-2 所示的两种异构体所处的动力学平衡中,在化学位移相对高场的一个甲基呈现出n(8a-1) :n(8a-2) 为1.0 :2.5 的比例;与之类似,化合物8b 和8e 两种异构体的比例为1.0 : 2.5 和1.0 : 4.0 (图2);所分离得到的E-构型产物9 也为两种互变异构体的混合物,比例都在1.0 : 1.6~1.0 : 2.0,苄基的亚甲基部分为一个单峰和一个J= 16.5 Hz 左右的AB 四重峰 (图2),说明其中一个异构体9a-1 的两个亚甲基质子为等价质子,而另一个异构体9a-2 的两个亚甲基质子为不等价质子。类似Z-构型产物8 的分析和前文[23-24]有关E-构型产物的晶体X-射线衍射分析结果,推断异构体9a-2 的C=N 双键位于咪唑啉酮环外,由于空间位阻导致苄基所在的CN 单键旋转受阻,从而使苄基亚甲基上的两个质子不等价形成一个偶合常数较大的AB 系统,通过9a-1 的亚甲基单峰和9a-2 的AB 四重峰积分面积得到比例n(9a-1) :n(9a-2) 在1.0 : 1.6 左右;与之类似,化合物9b 和9e 两种异构体的比例均为1 : 2 (图2)。为了进一步证实上述推测的结论,对8a 的两种互变异构体8a-1 和8a-2,以及9a 的两种互变异构体9a-1 和9a-2 进行了量子化学计算。结果 (表1) 表明,异构体8a-1 和8a-2 能量非常接近,它们的能量差只有0.26 kJ/mol,9a-1 和9a-2 的能量差也只有2.04 kJ/mol,这一结果也说明了无论是中间体6 或7 反应后均可以得到8a~8f 和9a~9h 两类产物,且8a~8f 和9a~9h 两类产物在溶液中均以两种互变异构体形式存在的原因。由于两种互变异构体的含量接近,通过不同溶剂重结晶来获取8a~8f 和9a~9h 的单晶,以了解在固体中8a~8f 和9a~9h 的结构存在形式的尝试未能获得成功。
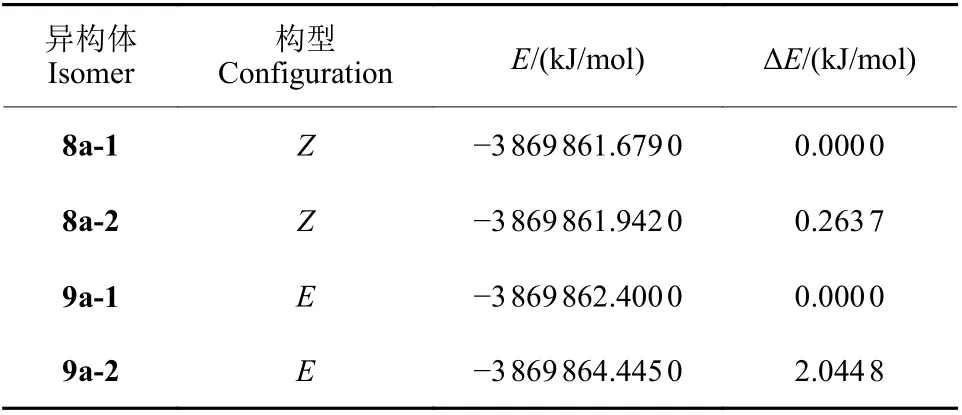
表1 化合物8a 和9a 互变异构体的量子化学计算结果Table 1 Quantum calculation results of compounds 8a and 9a tautomerism isomers
2.3 杀菌活性
2.3.1 离体杀菌活性 测试结果 (表2) 表明:在50 mg/L 下,具有E-构型的中间体5 和7 对5 种供试植物病原菌的抑制活性均好于Z-构型的中间体4 和6;具有E-构型的目标化合物9c、9d、9f和9h 对油菜菌核病菌的抑制活性也好于Z-构型的化合物8c、8d、8f 和8h。表明该类E-构型化合物的抑菌活性明显好于Z-构型化合物,说明5 位双键的构型对该类化合物的离体抑菌活性有显著影响。
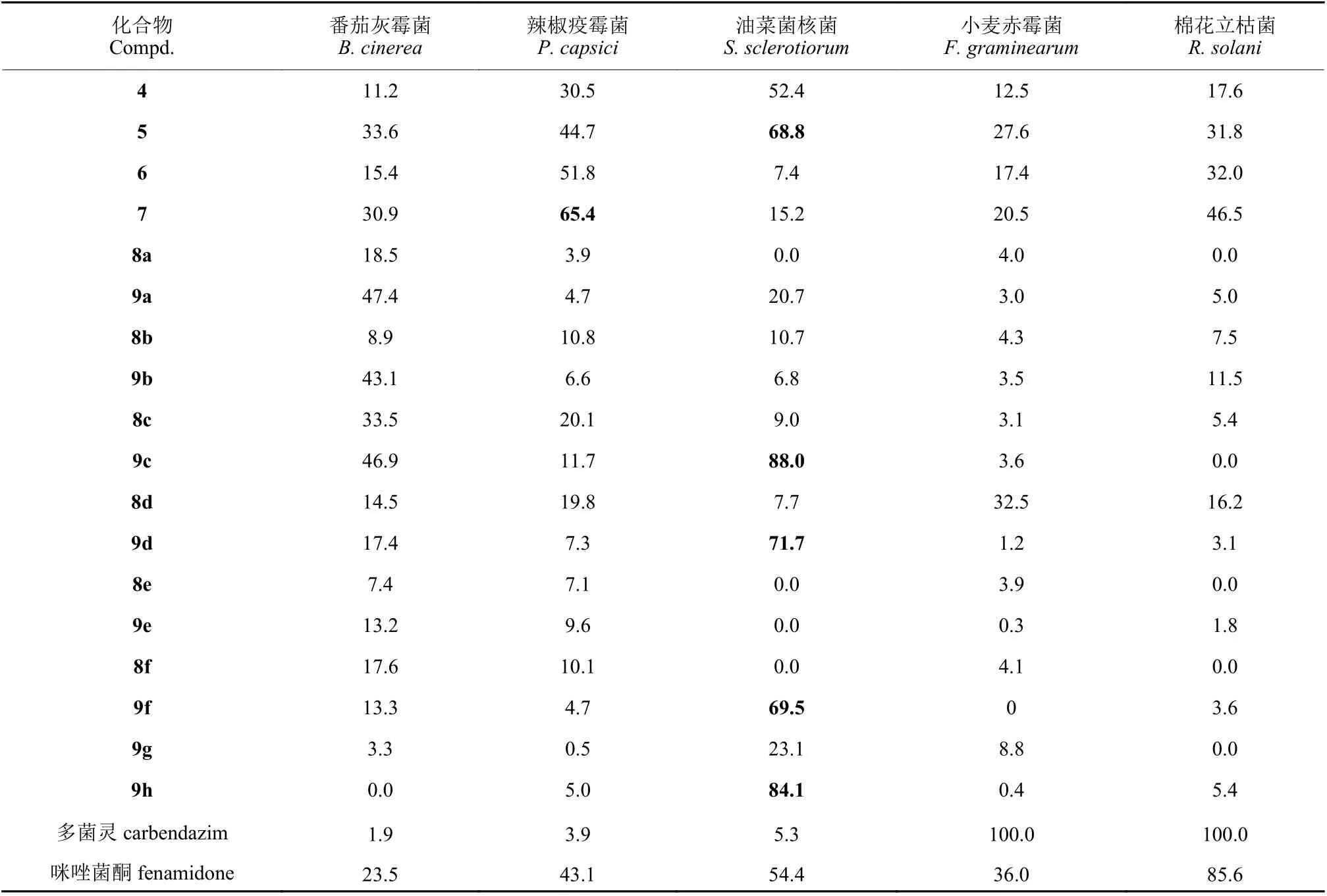
表2 目标化合物的离体杀菌活性 (抑制率/%, 50 mg/L)Table 2 The in vitro fungicidal activities of target compounds (inhibition rate/%, 50 mg/L)
化合物9c、9d 和9h 对油菜菌核病菌具有显著的抑制效果,在50 mg/L 下抑制率大于70%,抑制效果优于对照药剂咪唑菌酮,其余化合物对供试病原菌的抑制效果相对较差。毒力测试结果(表3) 显示,化合物9c、9d 和9h 对油菜菌核病菌的EC50值分别为14.3、21.8 和21.1 mg/L,显著优于对照药剂咪唑菌酮 (EC50= 46.8 mg/L)。分析该类化合物的化学结构和杀菌活性结果发现,当2 位苯胺上的取代基为4-OCH3、4-C(CH3)3和4-F时,对油菜菌核病菌的抑制效果较好,而4 位的CH3、Br、CF3和3 位的F 取代均导致抑菌活性显著降低。与本课题组前期所合成的3-位无苄基取代的2-氨基咪唑啉-4-酮系列化合物相比[23],在相同质量浓度下,9c、9f 和9h 对油菜菌核病菌的抑制率为88.0%、69.5%和84.1%,比相应的3-位无苄基取代的3 个化合物的抑制率 (46.8%、21.2%和49.9%) 有了明显提高[23],证明苄基的引入确实可以提高该类化合物的抑菌活性。
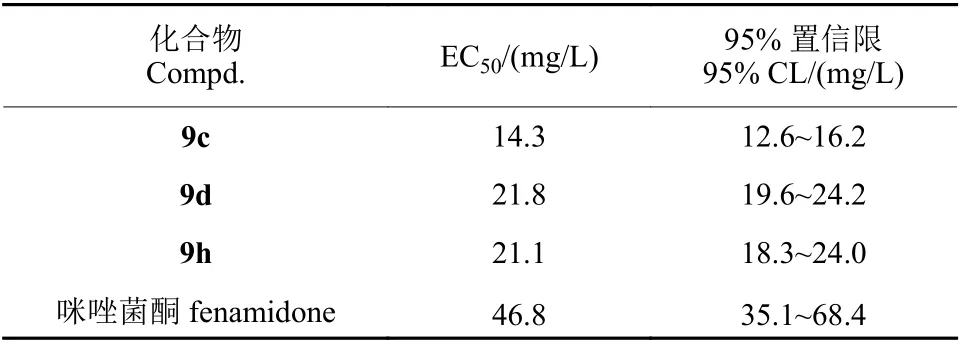
表3 部分化合物对油菜菌核病菌的EC50 值Table 3 EC50 values of some compounds against S. sclerotiorum
2.3.2 活体杀菌活性 测试结果 (表4) 表明:在400 mg/L 剂量下,中间体化合物4 和5 对于黄瓜霜霉病、小麦白粉病和玉米锈病均没有明显的防治效果,而硫酮甲基化后所得到的化合物6 和7 对黄瓜霜霉病显示一定的防治效果。用取代苯胺对中间体6 和7 进一步衍生化后的目标化合物,大部分对黄瓜霜霉病都表现出较好的防治效果,其中化合物8a、9c 的防治效果最好,均达到了80%,化合物9c 和8d 对于小麦白粉病也有较好的防治效果,防效分别为85%和80%,只有化合物9f 对玉米锈病显示出40%的防效。分析活体生测结果发现,当无取代和对位为给电子基团 (如t-C4H9、-OCH3) 取代苯胺时的化合物对黄瓜霜霉病具有一定的防治效果,而吸电子取代基或对位较弱给电子取代基存在时,化合物的防效明显下降。
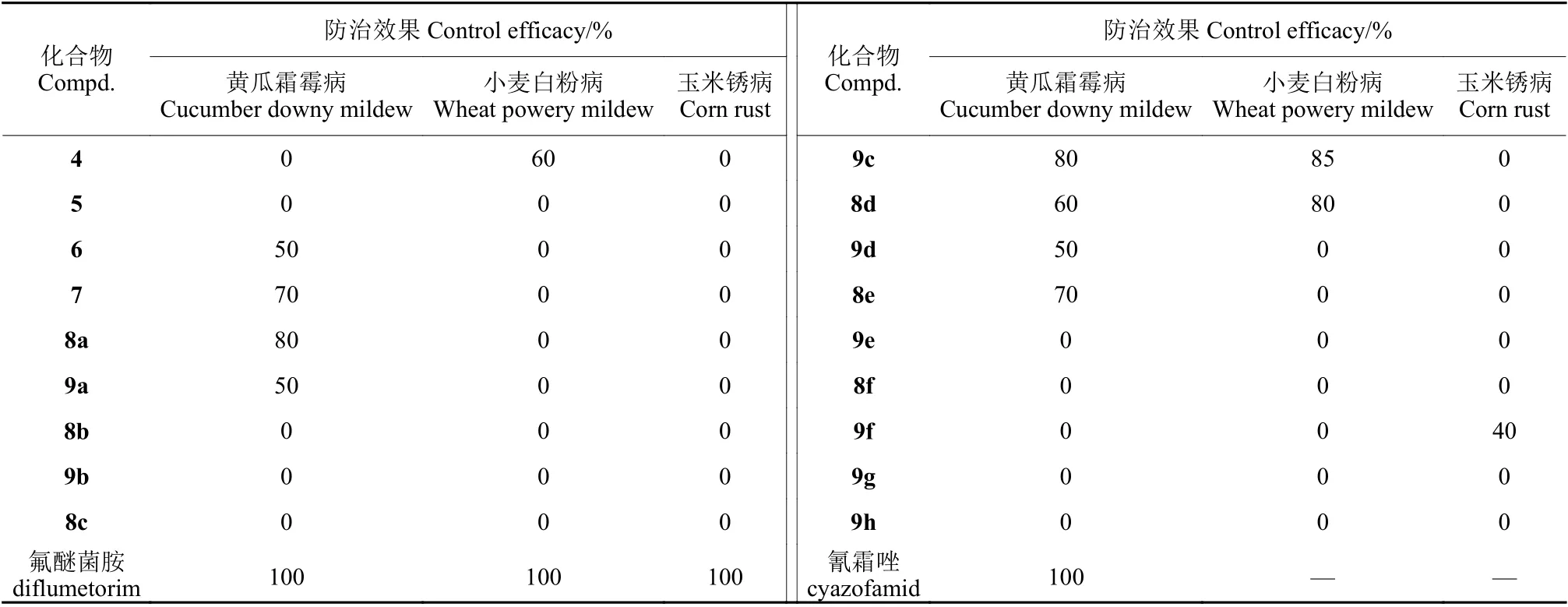
表4 目标化合物的活体杀菌活性 (400 mg/L)Table 4 The in vivo fungicidal activity of target compounds (400 mg/L)
离体和活体杀菌结果表明,采用对叔丁基苯胺和对甲氧基苯胺进行衍生化所得到的目标化合物活性最好,特别是 (E)-3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-(4-甲氧基苯基) 氨基咪唑啉-4-酮 (9c) 对于油菜菌核病菌、黄瓜霜霉病和小麦白粉病都具有较好的抑制效果,是值得进一步优化的先导结构。与离体抑菌活性不同,5 位双键的构型对该类化合物的活体杀菌活性没有显著影响,可能与多种环境因素有关,其具体原因尚待进一步探讨。
3 结论
采用活性亚结构拼接原理将螺环丁烯内酯和3-苄基咪唑啉酮进行拼接,设计并合成了一系列未见文献报道的3-苄基-5-(1-(4-甲基-2-氧代-1-氧杂螺[4,5]癸-3-烯-3-基) 亚乙基)-2-氨基咪唑啉-4-酮类化合物,并首次分离得到了Z-构型的该类化合物。经HPLC 分析验证,发现Z-和E-构型化合物均可以在酸性条件下通过氮质子化发生构型转化为E-和Z-构型化合物。初步杀菌活性测试结果表明,化合物9c、9d、9h 对油菜菌核病菌具有较好的离体抑制效果,化合物9c 的EC50值为14.3 mg/L;在400 mg/L 的剂量下,化合物9c 对黄瓜霜霉病和小麦白粉病具有较好的防治效果,防效分别达到80%和85%。3-位苄基的引入改善了该系列化合物的生物活性,其中化合物9c 表现出较好的生物活性,具有进一步结构优化的研究价值。
