3-甲基邻苯二酚/糠胺型聚苯并噁嗪的制备与性能
2021-12-16赵玉棣王立通
陈 辰,陆 馨,赵玉棣,王立通,辛 忠,2
(1.华东理工大学化工学院上海市多相结构材料化学工程重点实验室,2.化学工程联合国家重点实验室,上海200237)
聚苯并噁嗪作为一种热固性树脂,具有耐高温、低介电常数、低吸水率及固化收缩率接近于零等优点[1~5].这些优异的性能使得聚苯并噁嗪在电子产业、金属防护和航天航空等领域受到了广泛的关注[6].由于苯并噁嗪通常以热固化的方式聚合,存在固化温度过高的限制.因此,如何降低苯并噁嗪的聚合温度成为该领域备受关注的问题之一.
通常采用向苯并噁嗪单体中加入外源催化剂[7,8]、将不同苯并噁嗪单体进行共聚[9~11]和在结构中引入功能性官能团[12~14]这3种途径来降低苯并噁嗪的固化温度.其中,根据苯并噁嗪丰富的分子设计灵活性,在单体中引入具有催化作用的基团便可以从自身结构的角度实现内源催化.Zhang等[15]采用2-萘酚、3-氨基吡咯和甲醛溶液制备了羟基取代苯并噁嗪单体(NN-pd-OH),其固化峰值温度为183℃.Hao等[16]以芹菜素和糠胺为原料合成了含酚羟基的生物基苯并噁嗪树脂(API-fa),其固化峰值温度为189℃.
随着对自然界探索的逐渐深入,人们发现贻贝通过分泌贻贝黏附蛋白(MAPs)能够牢固地黏附于各种基质上,而MAPs的黏附力主要来源于3,4-二羟基苯丙氨酸(DOPA)中的邻苯二酚结构[17~20].Higginson等[21]合成了具有邻苯二酚基团的苯并噁嗪单体,拉伸剪切强度测试表明,与双酚A型苯并噁嗪树脂(B-a)相比,其在铝基材上的剪切强度提高了5倍以上.He等[17]使用苯酚、邻苯二酚、间苯二酚、对苯二酚和苯胺分别合成了4种苯并噁嗪单体,发现邻苯二酚型聚苯并噁嗪(PBOZ-ca)的拉伸剪切强度高于其它3种聚苯并噁嗪,表明邻苯二酚结构对于苯并噁嗪树脂的黏附性能具有重要影响.
本文设计合成了含有酚羟基的3-甲基邻苯二酚/糠胺型苯并噁嗪(M-f)单体,探究了酚羟基对于苯并噁嗪的开环聚合温度和黏附性能的影响,并合成间甲酚/糠胺型苯并噁嗪(MC-f)单体作为对照.通过差示扫描量热法(DSC)研究了2种苯并噁嗪单体的固化行为,采用Kissinger法和Ozawa法计算其表观活化能(Ea),采用热重分析仪(TG)研究了2种聚合物的热稳定性,在铝片和钢片上通过拉伸剪切强度测试考察了聚苯并噁嗪对金属的黏附性能.
1 实验部分
1.1 试剂与仪器
3-甲基邻苯二酚、间甲酚和糠胺,分析纯,上海麦克林生化科技有限公司;多聚甲醛和氯仿,分析纯,上海凌峰化学试剂有限公司.
Nicolet iS10型傅里叶变换红外光谱仪(FTIR),美国Nicolet公司;Ascend 600型核磁共振波谱仪(NMR),美国Varian公司,测试溶剂均为氘代氯仿,标准物为四甲基硅烷(TMS);DSC3+型差示扫描量热仪(DSC),瑞士Mettler-Toledo公司;Q500型热重分析仪(TG),美国TA仪器公司;BX51-P型光学显微镜,日本Olympus公司;GDXA40/150型微机控制电子万能试验机,美特斯工业系统有限公司.
1.2 M-f和MC-f的合成
采用3-甲基邻苯二酚、多聚甲醛和糠胺合成苯并噁嗪单体,反应式如Scheme 1所示.首先将10 mmol多聚甲醛和10 mmol糠胺加入三口烧瓶中,升温至80℃搅拌反应2 h;将10 mmol 3-甲基邻苯二酚、10 mmol多聚甲醛和20 mL氯仿加到烧瓶中,搅拌并回流反应;反应完毕后,待产物冷却至室温,用去离子水洗涤3次;将水洗后的溶液加入正己烷中,过滤后除去溶剂,冷冻干燥,得到M-f白色粉末,收率为62.9%.以间甲酚、多聚甲醛和糠胺为原料反应合成苯并噁嗪单体(Scheme 1).首先将150 mmol多聚甲醛和75 mmol糠胺加入三口烧瓶中,加入10 mL甲苯后室温搅拌1 h;再将75 mmol间甲酚溶于20 mL甲苯中,加入三口烧瓶中,搅拌并回流反应6 h;反应完毕后冷却至室温,对产物进行多次重结晶,冷冻干燥后得到MC-f白色晶体,收率为76.8%.

Scheme 1 Synthesis route of M⁃f and MC⁃f monomers
1.3 测试与表征
1.3.1 苯并噁嗪单体的热固化行为研究通过原位红外(in-situFTIR)和差示扫描量热仪(DSC)对M-f和MC-f苯并噁嗪单体的热固化过程进行研究.将M-f和MC-f单体与溴化钾充分研磨,压片后置于红外热台中,加热速率为5℃/min,每隔10℃采集1次谱图.此外,将M-f分别在120℃或140℃固化2 h,得到聚苯并噁嗪PM-f-120℃和PM-f-140℃;将MC-f单体分别在180℃或200℃下固化2 h,得到聚苯并噁嗪PMC-f-180℃和PMC-f-200℃,用于后续的DSC测试.采用DSC分析苯并噁嗪单体的非等温固化行为,测试条件:在N2气氛下,称取3~5 mg样品,升温速率为10℃/min,温度范围为40℃到300℃.
1.3.2 聚苯并噁嗪的热稳定性测试使用热重分析仪对聚苯并噁嗪PM-f-140℃及PMC-f-200℃的热稳定性进行研究.取约10 mg聚苯并噁嗪,在N2气保护下,以10℃/min的升温速率从40℃升温到800℃.
1.3.3 聚苯并噁嗪的黏附性能测试通过拉伸剪切强度测试考察聚苯并噁嗪对于金属基材的黏附性能,根据ASTM D1002-10标准制备样品.首先对铝片和经过打磨的低碳钢片(2.50 cm×10.00 cm)进行溶剂脱脂处理,用耐热胶带将金属基材端部的2.50 cm×1.00 cm区域隔离.然后将苯并噁嗪单体溶于溶剂中,滴在隔离区域,挥发溶剂后在真空干燥烘箱中干燥1 h;取下耐热胶带后,将两片基材搭接,并用夹子夹持;将M-f和MC-f涂覆的金属基材先在100℃加热1 h,然后再分别在140℃或200℃下固化2 h,缓慢冷却至室温后,从黏合区域边缘去除多余的聚苯并噁嗪,分别命名为PM-f和PMC-f.使用万能试验机对制备的样品进行拉伸剪切强度测试,以5 mm/min的恒定速度拉伸试样,直至试样失效.使用光学显微镜观察样品失效接头处的形貌.
2 结果与讨论
2.1 苯并噁嗪单体的结构表征
图1 是M-f和MC-f单体的FTIR谱图.对于M-f单体,950 cm−1处为噁嗪环的特征吸收峰,1012 cm−1处为C—O—C键的对称伸缩振动峰,1257 cm−1处为C—O—C键的反对称伸缩振动峰,1155 cm−1处为噁嗪环上C—N—C的对称伸缩振动峰,737和1588 cm−1处的吸收峰对应于呋喃环的特征吸收峰,M-f四取代的Ar—H振动吸收峰出现在1471 cm−1处,在2921和2851 cm−1处的吸收峰归属于噁嗪环上及与呋喃环连接处的亚甲基;另外,在3407 cm−1处的吸收峰表明M-f单体中存在酚羟基.而MC-f单体的噁嗪环特征吸收峰出现在956 cm−1处,C—O—C键的反对称伸缩振动峰出现在1242 cm−1处,三取代Ar—H振动吸收峰出现在1505 cm−1处.
图2 给出M-f和MC-f单体的1H NMR谱图.对于M-f单体,在δ3.92和4.95处出现的2个共振峰分别归属于噁嗪环的Ar—CH2—N和O—CH2—N;δ2.23处的单峰归属于CH3质子;δ7.26处的吸收峰为溶剂峰(CDCl3);δ3.99处的吸收峰为糠胺结构中CH2的吸收峰;δ7.42,6.34和6.25处的吸收峰归属于呋喃环中的质子;δ6.44和6.65处的吸收峰归属于苯环上的芳族质子.对于MC-f单体,δ3.91和4.86处的吸收峰归属于Ar—CH2—N和O—CH2—N的CH2;δ3.97处为呋喃环和噁嗪环相连的CH2信号;δ7.40,6.32和6.24处的吸收峰归属于呋喃环中的质子;δ6.83,6.71和6.64处的吸收峰归属于苯环上质子;δ2.28处的吸收峰归属于与苯环相连的CH3质子.
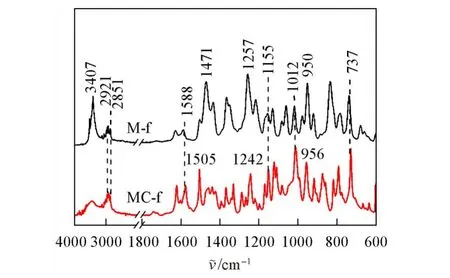
Fig.1 FTIR spectra of M⁃f and MC⁃f

Fig.2 1H NMR spectra of M⁃f and MC⁃f

Fig.3 13C NMR spectra of M⁃f and MC⁃f
图3 为M-f和MC-f单体的13C NMR谱图.M-f单体在δ82.35和48.92处出现的2个共振峰分别为噁嗪环的O—CH2—N和Ar—CH2—N的碳峰;δ109.06~151.48之间的吸收峰是呋喃环和苯环上的碳峰;δ48.12处为连接呋喃环的—CH2—N的碳峰,δ15.33处的吸收峰为甲基的碳峰.MC-f单体在δ81.77和49.32处的吸收峰归属于噁嗪环上的O—CH2—N和Ar—CH2—N的碳原子.
2.2 苯并噁嗪单体的热固化行为
图4 为M-f和MC-f的DSC谱图.从图4可以看出,在10℃/min的升温速率下,M-f单体和MC-f单体的熔点分别是97℃和80℃.含有酚羟基的M-f单体的固化起始温度(Tonset)为160℃,固化峰值温度(Tp)为172℃;而MC-f单体的Tonset为233℃,Tp为244℃.M-f的Tp比MC-f低72℃,表明酚羟基的引入有利于降低苯并噁嗪的固化峰值温度.M-f单体在具有较低固化温度的同时还有近70℃的加工窗口,这有利于M-f的实际应用.
采用非等温DSC对苯并噁嗪的开环聚合动力学进行研究,加热速率分别为2,5,10,15和20℃/min,结果如图5所示.开环聚合过程的表观活化能(Ea,J/mol)通过Kissinger和Ozawa方法进行计算[22],计算公式分别如式(1)和式(2)所示:
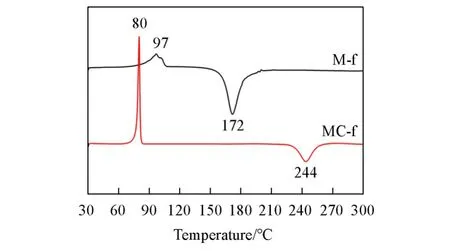
Fig.4 DSC curves of M⁃f and MC⁃f
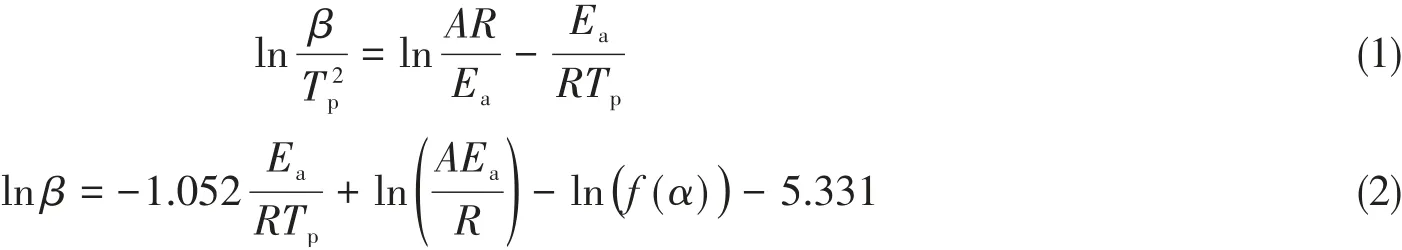
式中:β(K/min)为升温速率;Tp(K)为固化峰值温度;A(min−1)为指前因子;R(8.314 J·mol−1·K−1)为普适气体常量.分别以ln(β/T2p)和lnβ为因变量,(1/Tp)为自变量,拟合出直线.通过斜率就可以求出Ea,通过截距能够求出指前因子A.
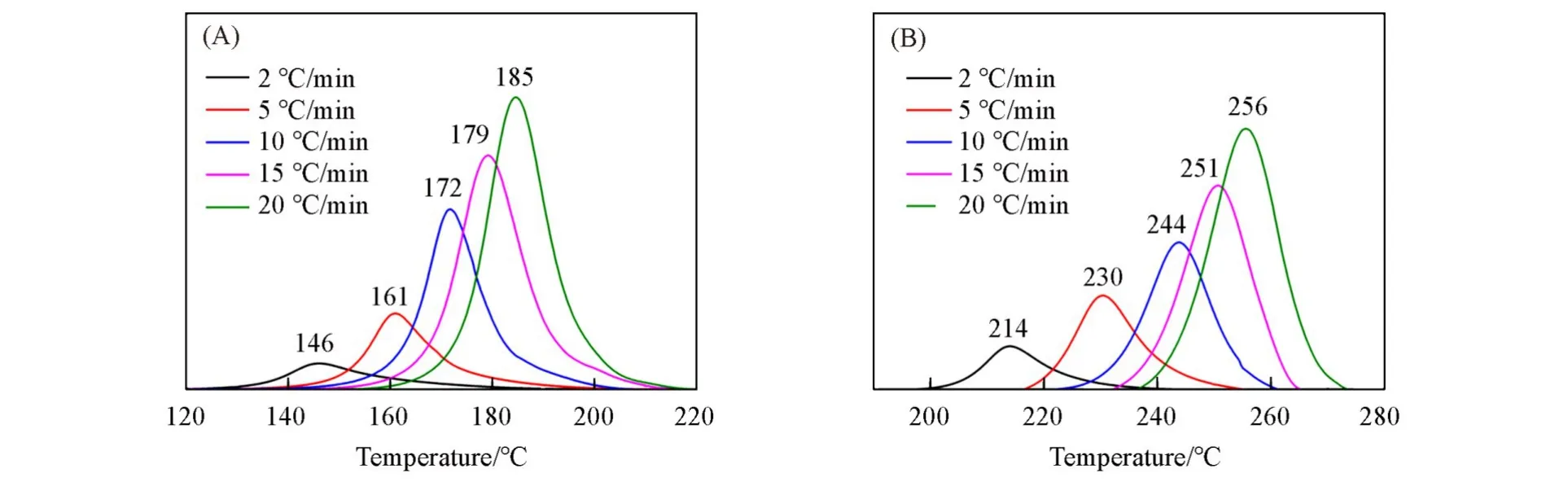
Fig.5 DSC curves of M⁃f(A)and MC⁃f(B)at various heating rates
根据Kissinger和Ozawa方法,分别以ln(β/T2p)和lnβ对1/Tp作图(图6).通过上述两种方法计算得到M-f的Ea分别为88.19和90.74 kJ/mol,而MC-f的Ea分别为107.25和109.85 kJ/mol.对于同种单体,分别采用两种方法计算出来的表观活化能十分接近,产生的差值可能是由于两种方法的假设不同.M-f开环聚合反应活化能相比MC-f低约20 kJ/mol,表明M-f比MC-f更易发生开环聚合反应,进一步说明酚羟基结构对于苯并噁嗪单体开环聚合具有促进作用.

Fig.6 Plots generated following the Kissinger(A)and Ozawa(B)methods for determination of the activation energy of M⁃f and MC⁃f
进一步通过原位红外光谱对于苯并噁嗪单体的固化行为进行了探究,图7为M-f和MC-f样品在加热过程中不同温度下的红外谱图.当温度达到180℃时,M-f苯并噁嗪在950 cm−1处噁嗪环的特征峰几乎消失.而对于MC-f,956 cm−1处的噁嗪环特征峰在温度达到230℃时基本消失.这两种结构的苯并噁嗪单体的FTIR结果与DSC分析结果一致,表明酚羟基的引入促进了单体的开环聚合.

Fig.7 In⁃situ FTIR spectra of M⁃f(A)and MC⁃f(B)at various temperatures with a constant heating rate of 5℃/min
将M-f单体分别在120℃或140℃下固化2 h得到PM-f-120℃和PM-f-140℃,将MC-f分别在180或200℃下固化2 h得到PMC-f-180℃和PMC-f-200℃,并对上述样品进行FTIR和DSC表征.从图8可以看出,MC-f单体在180℃下固化2 h后,DSC曲线中仍存在放热峰,而在200℃下固化后则没有放热峰出现,表明MC-f单体需要在200℃下固化2 h才得以完全固化.而PM-f单体在140℃固化2 h后放热峰就已经消失,证明其在较低的温度下即可完全固化.
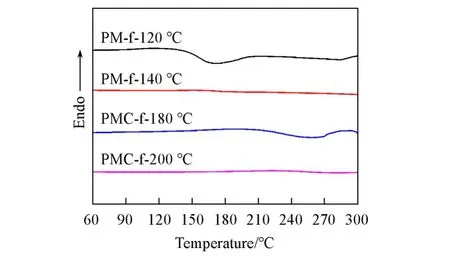
Fig.8 DSC curves of PM⁃f⁃120℃,PM⁃f⁃140℃,PMC⁃f⁃180℃and PMC⁃f⁃200℃
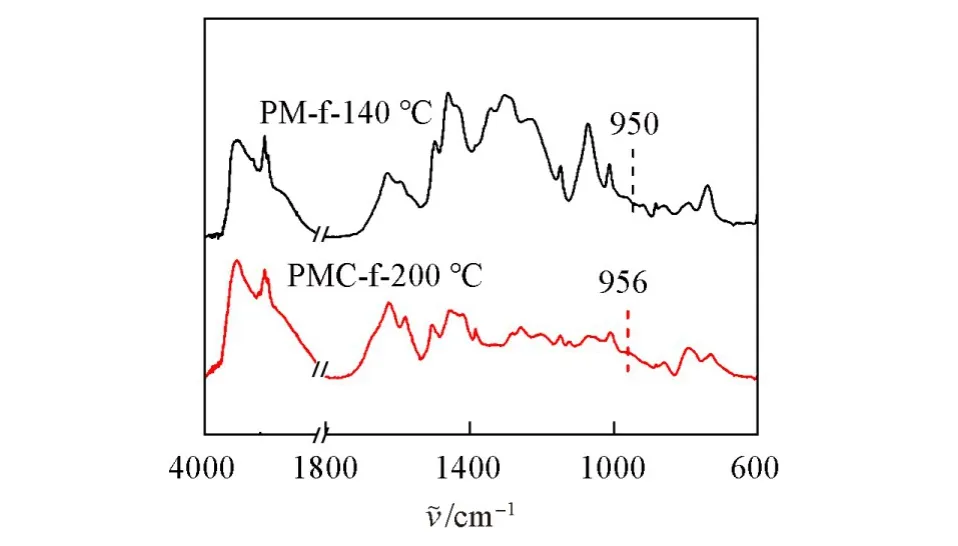
Fig.9 FTIR spectra of PM⁃f⁃140℃and PMC⁃f⁃200℃
图9 为M-f和MC-f在不同温度下固化2 h后的FTIR图.M-f在950 cm−1处的噁嗪环特征峰在140℃下固化2 h后完全消失,MC-f在200℃下固化2 h后噁嗪环特征峰也消失不见,说明两种单体在这种条件下都能够完全开环固化.
图Scheme 2示出了M-f的热固化聚合机理.M-f单体中的酚羟基在加热条件下可以促进噁嗪环上C—O键的断裂,导致M-f的开环聚合温度低于MC-f单体.此外,由于O原子的对位C原子电子云密度增加,碳正离子可能与苯环上富电子的位置发生亲电取代反应;而呋喃基团也易于发生反应,从而会提供额外的交联位点参与苯并噁嗪的开环聚合反应.最终,开环聚合形成的PM-f分子结构中具有邻苯二酚结构.
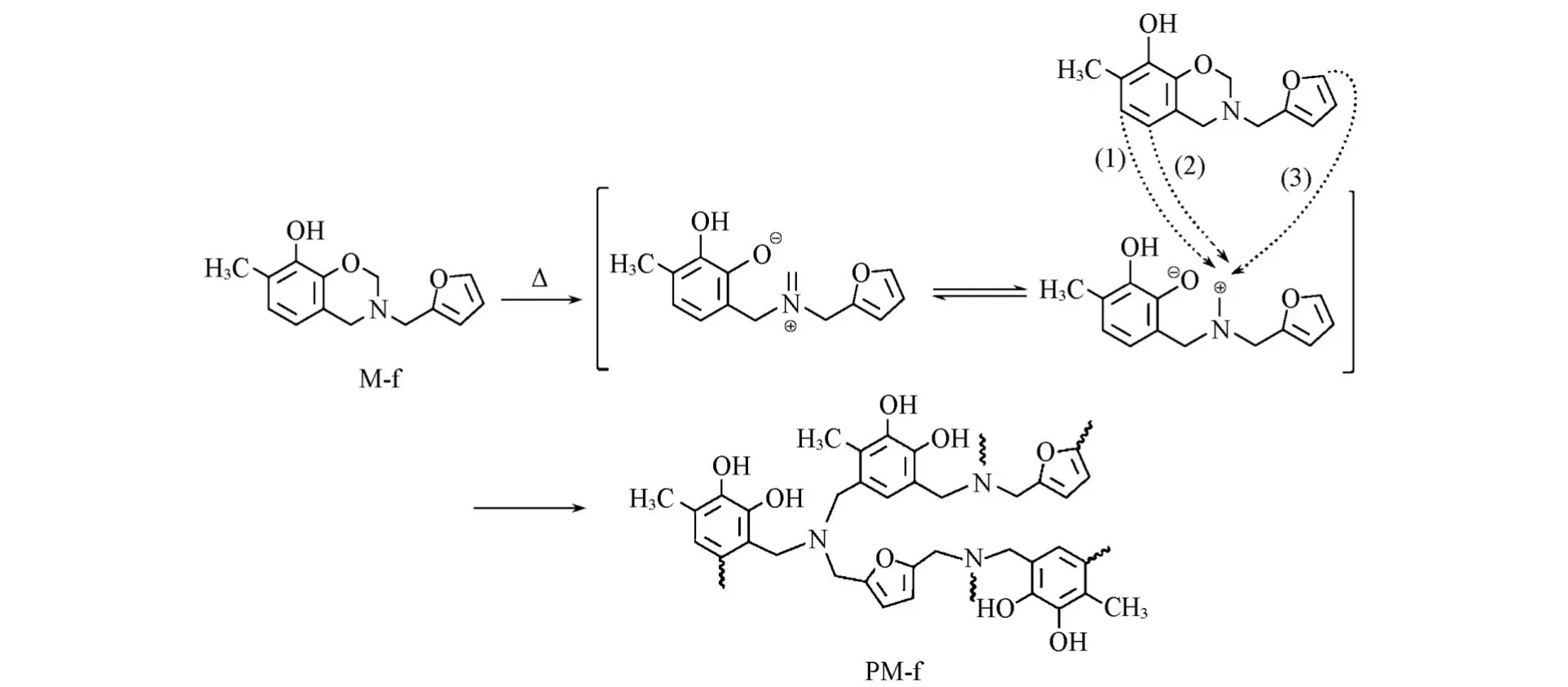
Scheme 2 Schematic diagram of polymerization of M⁃f
2.3 聚苯并噁嗪的热稳定性
图10 示出聚苯并噁嗪PM-f和PMC-f的TGA曲线.PM-f在250~380℃的温度范围内具有明显失重,而PMC-f则在350~480℃之间具有显著的质量损失,这主要是由于聚苯并噁嗪中Mannich桥结构发生破坏.PM-f的T1%,T5%和T10%(226,279和306℃)均低于PMC-f(277,342和378℃),这可能是由于PM-f结构中酚羟基的存在使得Mannich桥上的C—N键更易发生断裂并释放出Schiff碱,从而造成前期的失重.随着温度的进一步升高,PM-f和PMC-f开始发生碳化过程,最终在800℃下的残碳率分别为51.4%和45.7%,这归因于两者结构中的呋喃环部分增加了聚苯并噁嗪的热稳定性.其中,PM-f中的酚羟基能够增加聚苯并噁嗪的交联密度,使其残碳率高于PMC-f.

Fig.10 TGA curves of PM⁃f and PMC⁃f

Fig.11 Lap shear strength of PM⁃f and PMC⁃f
2.4 聚苯并噁嗪的黏附性能
M-f单体在开环聚合后会形成具有邻苯二酚结构的聚苯并噁嗪,这种仿贻贝结构可以通过与金属基材表面的化学作用提高黏附性能.PM-f和PMC-f的拉伸剪切强度如图11所示.PM-f的拉伸剪切强度在铝和低碳钢基材上分别达到2.53和3.09 MPa,而PMC-f在铝和钢基材上的拉伸剪切强度分别为1.22和1.67 MPa.
此外,我们还研究了接头处黏附性的失效模式.当聚苯并噁嗪与基材之间的黏附界面发生黏附失效时,即为黏附破坏.当接头失效是由聚苯并噁嗪本体发生破坏所致时,则为内聚破坏.当在失效的接头中同时观察到黏附和内聚破坏的区域时,则为混合破坏.图12为PMC-f和PM-f分别在铝和低碳钢基材上失效接头的光学显微镜图片.PMC-f和PM-f在铝基材上的接头主要表现为黏附失效,而在低碳钢基材上均表现为混合失效,表明这两种聚苯并噁嗪与低碳钢表面具有更强的相互作用.这是由于低碳钢基材在搭接之前经过了砂纸打磨的预处理步骤,粗糙的低碳钢表面增加了基材与聚苯并噁嗪之间的黏附面积,从而表现出比铝基材更强的黏附性.

Fig.12 Optical images of lap joints after shear testing of PMC⁃f on aluminum sheet(A),PMC⁃f on mild steel(B),PM⁃f on aluminum sheet(C)and PM⁃f on mild steel(D)
拉伸剪切强度测试和失效接头的表面形貌观察结果表明,PM-f和PMC-f均对金属基材表现出一定的黏附性能,而PM-f对于铝片和钢片的黏附性能均大于PMC-f,说明邻苯二酚结构对于改善聚苯并噁嗪的黏附性能起到了重要作用.聚苯并噁嗪的分子结构中含有大量的酚羟基,对金属表现出良好的黏附性能.PM-f中的邻苯二酚基团可以与金属基材表面原子之间形成配位键,从而使PM-f和金属基材之间形成更强的相互作用力[17].
3 结 论
设计合成了含有酚羟基的3-甲基邻苯二酚/糠胺型苯并噁嗪(M-f),并合成间甲酚/糠胺型苯并噁嗪(MC-f)单体作为对照.DSC结果表明,M-f具有较低的固化温度,其放热峰值温度(Tp)为172℃,比MC-f低72℃.TG测试结果表明,聚苯并噁嗪PM-f在800℃下的残碳率高达51.4%,证明其具有良好的热稳定性.此外,在铝片和低碳钢片上通过拉伸剪切强度测试探究了M-f和MC-f的黏附性能.测试结果表明,含有邻苯二酚结构的PM-f对于铝和低碳钢基材具有良好的黏附性能,拉伸剪切强度分别达到2.53和3.09 MPa,均高于PMC-f.总体而言,含有酚羟基的苯并噁嗪具有较低的固化温度,并赋予苯并噁嗪树脂良好的热固化性能及黏附性能.
