反溶剂法快速合成高效发光二维锡卤钙钛矿材料
2021-12-16邓正涛
刘 瑶,邓正涛
(南京大学现代工程与应用科学学院,南京210023)
金属卤化物钙钛矿半导体材料具有许多优异的光电性能,如高载流子迁移率、高消光系数、可调的吸收和发射光谱、窄的半峰宽、高荧光量子产率(PLQY)及高缺陷容忍度等,并且其溶液合成工艺简单[1],在太阳能电池、发光二极管(LED)、荧光太阳能聚光器、光电探测器、激光和液晶显示器等领域展现出巨大的应用潜力[2~4].目前,被广泛研究的铅卤钙钛矿太阳能电池(PSCs)的光电转换效率已达到25.5%,且基于铅卤钙钛矿的LED的外量子效率也超过20%[5].然而,铅卤钙钛矿材料中都存在重金属元素,并且铅对人体健康和生态环境都会造成严重的威胁.因此,研究者一直致力于开发光电性质优异的非铅卤化物钙钛矿材料[6].锡与铅是同主族相邻元素,与铅具有相同的价态和相近的离子半径,是最常见的铅的替代品.锡卤钙钛矿具有低毒性、合适的带隙、高吸光系数和高载流子迁移率等优异的特性.但是其结构中的固有缺陷和二价锡离子的氧化(Sn2+→Sn4+)会导致晶体自掺杂效应,从而严重影响其器件性能[7,8].目前用于改善锡卤钙钛矿光电性能和稳定性的策略主要有4种:(1)添加草酸或次磷酸等还原剂来抑制Sn2+的氧化;(2)降低锡基钙钛矿的维度(3D→2D);(3)采用四价锡取代二价锡;(4)离子掺杂.其中通过引入长链有机胺阳离子作为A位合成2D结构的锡卤钙钛矿材料是最关键的举措.相比3D结构,2D锡卤钙钛矿往往具有更加优异的光学性能和稳定性,这是由于具有大空间位阻和疏水性的有机阳离子能有效提高[SnX6]−发光中心对水和氧气的耐受程度[9].
近年来,关于2D锡卤钙钛矿的研究已取得一定进展,合成方法主要包括反溶剂法和热注射法[10].Haque课题组[8]制备了2D(PEA)2SnI4(PEA=苯乙胺)薄膜,并将其应用于LED的发射层.相比传统的3D CH3NH3SnI3钙钛矿材料,该薄膜具有更优越的光学性质和空气稳定性.Ning课题组[9]采用反溶剂法合成了中空结构的2D HMD3SnBr8钙钛矿晶体(HMD=己二胺),他们将HMDBr2和SnBr2按照一定的化学计量比溶解于DMF溶液中,随后将该前驱体溶液转移至二氯甲烷中,得到了发明亮黄色光的HMD3Sn-Br8晶体,其PLQY高达86%,与商用蓝色荧光粉混合可制备得到白光LED灯.Rogach课题组[11]采用高温热注射法合成了发明亮橙色光的2D(C18H35NH3)2SnBr4钙钛矿薄膜(PLQY=68%),并用其制备了具有优异性能的电致发光LED,其亮度达到350 cd/m2.然而由于合成过程中需要使用昂贵且毒性 高的三正辛基膦试剂,不利于大规模生产.Lu等[12]以TEAI和SnI2作为原料,采用反溶剂法合成了2D(TEA)2SnI4薄膜(TEA=2-噻吩乙胺),并实现了基于该薄膜的纯红色电致发光LED的制备.
最近,本课题组[13]首次在室温酸辅助水溶液条件下合成了PLQY接近100%的2D(C8H17NH3)2SnBr4钙钛矿,该材料表现出极好的空气稳定性,在一定温度和湿度的大气环境下储存240 d,其PLQY基本保持不变.我们[14]还采用高温热注射法合成了2D(RNH3)2SnX4钙钛矿,虽然采用该方法合成的样品尺寸、形貌更加均一,且稳定性优异,但合成条件严苛,反应较复杂.
反溶剂法在常温常压条件下即可进行,操作简便,反应快速,为材料的大规模生产提供了可能.其原理及主要的反应过程为:反应物首先溶解于良溶剂[N,N-二甲基甲酰胺(DMF)、二甲基亚砜等]中形成前驱体溶液,当将少量前驱体溶液加入到适量不良溶剂(二氯甲烷、丙酮等)中时,反应体系迅速变为过饱和溶液,过饱和度驱动反应产物逐渐成核、结晶和生长.本课题组[15]之前报道了全无机铯铅溴钙钛矿量子点的反溶剂法可控制备.
本文采用反溶剂法快速合成了一系列高效发光的2D(RNH3)2SnX4(X=Br,I)钙钛矿,其中RNH3+表示长链有机胺阳离子,分别为丁胺阳离子(C4H9NH3+)和辛胺阳离子(C8H17NH3+).我们的工作具有以下的优势和意义:(1)首次通过反溶剂法合成了光学性能优异的2D(C4H9NH3)2SnBr4和(C8H17NH3)2SnBr4钙钛矿,且其PLQY分别达到98.5%和88%;(2)(C8H17NH3)2SnBr4具有良好的空气稳定性;(3)通过调节卤素含量,可将(C8H17NH3)2SnX4的荧光发射从黄色光调至橙红色光.
1 实验部分
1.1 试剂与仪器
正丁胺、正辛胺、二氯甲烷和碘化锌(ZnI2)均购于上海阿拉丁试剂有限公司;氢溴酸、正己烷和三甲基碘硅烷(TMSI)均购于上海麦克林生化科技有限公司;DMF购于西陇科学股份有限公司;溴化亚锡(SnBr2)购于阿法埃莎公司.
Bruker AXS D8型粉末X射线衍射仪(PXRD,德国Bruker公司);NEXUS870型傅里叶变换红外光谱仪(FTIR,美国Nicolet公司);UV-3600 Plus型紫外-可见-近红外吸收光谱仪(UV-Vis-NIR,日本Shimadzu公司);ULTRA55型扫描电子显微镜(SEM,德国Zeiss公司);PTI QuantaMaster 400型荧光光谱仪(PL,日本Horiba公司);Fls920型低温时间分辨荧光光谱仪(英国Edinberge公司);PHI 5000 VersaProbe型X射线光电子能谱仪(XPS,日本Ulvac-Phi株式会社);INCA X-MAX型能谱仪(EDS,英国牛津仪器公司);UNIQUE R10型超纯水系统(锐思捷科学仪器有限公司).
1.2 实验过程
1.2.1 辛胺溴盐(C8H17NH3Br)/丁胺溴盐(C4H9NH3Br)的制备将一定量的正辛胺或正丁胺与氢溴酸反应,经旋转蒸发、抽滤、冷冻干燥等步骤,得到提纯的辛胺溴盐或丁胺溴盐.
1.2.2 (C8H17NH3)2SnBr4的制备称取过量的辛胺溴盐固体和SnBr2粉末溶解在DMF溶液中,超声振荡约5 min,得到澄清透明的前驱体溶液.然后将该溶液转移至二氯甲烷中,体系逐渐呈白色浑浊,以10000 r/min转速离心5 min得到白色(C8H17NH3)2SnBr4钙钛矿.
1.2.3 (C4H9NH3)2SnBr4的制备将过量的丁胺溴盐固体和SnBr2粉末溶解在DMF溶液中,后续实验步骤与1.2.2节一致.
1.2.4 (C8H17NH3)2SnX4(X=Br,6Br/I,4Br/I)的原位合成将一定量的TMSI滴加至辛胺溴盐和SnBr2的混合物中,然后向其中加入DMF溶液,最后将该混合溶液转移至二氯甲烷中,超声振荡、离心洗涤得到白色沉淀.改变TMSI的含量即可得到不同Br/I比例的(C8H17NH3)2SnX4钙钛矿.
1.2.5 (C8H17NH3)2SnX4的离子交换反应合成将制得的(C8H17NH3)2SnBr4固体重新分散在正己烷溶液中,向其中加入不同量的ZnI2固体,超声混合均匀,即可得到不同Br/I比例的(C8H17NH3)2SnX4钙钛矿.
2 结果与讨论
2.1 (RNH3)2SnBr4的结构、形貌与组成表征
为了简便起见,在下文讨论中采用C4,C8分别代替(C4H9NH3)2SnBr4和(C8H17NH3)2SnBr4钙钛矿.采用XRD对样品的晶体结构进行表征,图1(A)为样品的XRD谱图.可以看到,C4和C8都具有周期性排布的衍射峰,符合2D钙钛矿的特征[15,16].经布拉格方程2dsinθ=nλ计算,C4和C8的晶面间距(d)值分别为1.51 nm和2.46 nm[17].在2D(RNH3)2SnBr4钙钛矿的晶体结构中,[SnBr6]−呈层状排列,有机胺链作为保护层将[SnBr6]−八面体间隔开[14].
采用FTIR对样品的分子结构进行了表征,图1(B)和(C)分别为C4、C8与所有反应原料的FTIR谱图,其中位于1000~1750 cm−1和2750~3000 cm−1的所有特征峰都归属于基本的胺链结构[18].可以看到,反应后丁胺溴和辛胺溴的N—H(—NH3+)伸缩振动峰位置发生了明显改变,分别由3438 cm−1和3370 cm−1移动至3110 cm−1(C4)和3112 cm−1(C8),这是由于反应后—NH3+与[SnBr6]4−进行了连接(图中红色虚线圆标记).其它对应的特征峰如下:C—H的对称伸缩振动峰:2861和2925 cm−1;N—H的弯曲振动峰:1579 cm−1;C—N的伸缩振动峰:1097 cm−1;—CH3弯曲振动峰:1477 cm−1.综合FTIR图谱分析,可以初步判定丁胺阳离子和辛胺阳离子是(RNH3)2SnBr4钙钛矿的主要有机成分.
图2 为C8的XPS谱图.图2(A)表明C8中含有C,N,Sn,Br等元素,而532 eV处的O1s峰应归属于化学吸附的O元素,这可能是由于在测试过程中样品表面引入了杂质.图2(B)中位于401.4 eV处的峰归属于N1s;由于具有疏水性和大空间位阻的有机胺阳离子作为A位,(C8H17NH3)2SnBr4钙钛矿具有优异的空气稳定性,Sn2+难以氧化成Sn4+.因此,图2(C)中位于486.2和494.5 eV处的峰归属于图2 (D)显示Br3d的峰位置在68.0 eV处.根据峰面积积分,计算出C8中n(N)∶n(Sn)∶n(Br)=4.5∶1.0∶5.5,检测到的总氮元素含量来源于辛胺阳离子,其中一部分按化学计量比参与合成(C8H17NH3)2SnBr4钙钛矿,过量的未反应的部分则吸附在钙钛矿的表面起配体保护作用[13].
图3 (A1)~(A3)和(B1)~(B3)分别为C4和C8的SEM照片.可以观察到两者均具有2D层状结构,与XRD实验结果相吻合.其中C4和C8的形貌有所差异,C4的表面呈带状或者分支状,而C8的表面更为规整.通过EDS研究样品的元素分布情况,结果表明,N,Sn,Br元素在C4和C8样品中呈现均匀的分布[图3(C1)~(C3)和(D1)~(D3)].

Fig.2 Survey(A)and high resolution(B—D)X⁃ray photoelectron spectra of C8

Fig.3 Scanning electron microscopy images of C4(A1—A3),C8(B1—B3)and energy⁃dispersive X⁃ray spec⁃troscopy mapping images of C4(C1—C3)and C8(D1—D3)
2.2 (RNH3)2SnBr4的光学性质和稳定性
图4 (A1)和(A2)为C4和C8的己烷胶体溶液在室内正常光照(A1)和365 nm紫外光照射(A2)下的光学照片,可以看到C4和C8均具有强黄色荧光.图4(B1)和(B2)为C4和C8的紫外吸收光谱图(UV)、荧光发射光谱图和激发光谱图.C4和C8均表现出宽的荧光发射,半峰宽约为118 nm,发射峰分别位于598 nm和600 nm处.C4的激发峰和吸收峰位置分别为351 nm和360 nm,C8的激发峰和吸收峰位置分别为330 nm和341 nm.(αhν)2-hν关系曲线[图4(C1)和(C2)]表明,C4和C8均为直接带隙材料,经计算,其带隙(Eg)分别为3.246 eV(C4)和3.172 eV(C8).二者均具有大的斯托克斯位移,分别为238 nm(C4)和259 nm(C8).由于C4和C8的胺链长度不同,[SnBr6]−八面体发光中心将产生不同的瞬态弹性晶格畸变,导致二者的发射峰和吸收峰位置有所差异[20].

Fig.4 Photographs of the C4 and C8 colloidal suspension under room(A1)and UV light(A2),UV⁃absorp⁃tion(a),PL emission(b)and PL excitation(c)spectra of C4(B1)and C8(B2)and(αhυ)2⁃hυ plots of C4(C1)and C8(C2)

Fig.5 Absolute PLQYs of as⁃synthesized 2D C4(A)and C8(B)perovskites under different excitation wave⁃lengths and three⁃dimensional excitation⁃emission matrix fluorescence spectra of C4(C)and C8(D)
如图5(A)和(B)所示,C4和C8均具有激发波长依赖的PLQY,且分别在330和320 nm处达到其PLQY的最大值,分别为98.5%(C4)和88.0%(C8).另外,对样品进行了三维激发-发射矩阵荧光光谱(EEM)测试.由图5(C)和(D)可以看到,随激发峰位置从300 nm变化至400 nm,二者的发射峰位置几乎不变,说明样品中没有杂质或者附加能级的存在[13].
图6 (A)和(B)分别为C4和C8的荧光寿命衰减曲线.用单指数函数对其进行拟合,发现C4和C8均具有长的荧光衰减寿命(τavg),分别为5.37 μs(C4)和5.51 μs(C8),拟合度R2>0.99.为探究样品的τavg与PLQY之间的内在联系,分别计算了C4和C8的辐射衰减速率(kr=PLQY/τavg)、非辐射衰减速率[knr=(1/τavg)−kr]以及两者的比值(kr/knr).如表1所示,与C8相比,C4的kr/knr增大,kr占比更大,说明C4中的辐射复合通道多于非辐射复合通道,因而其PLQY大于C8[19].
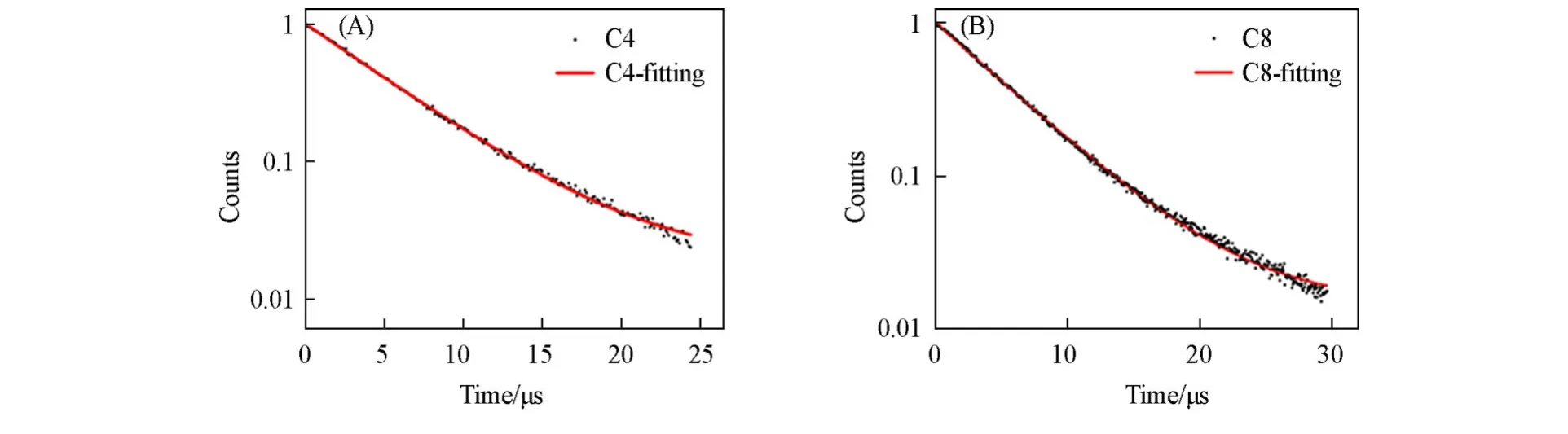
Fig.6 Time⁃resolved PL decays spectra of C4(A)and C8(B)

Table 1 kr,knr and kr/knr of C4 and C8
综上所述,C4和C8钙钛矿均具有宽的荧光发射、大的斯托克斯位移和长的荧光衰减寿命,这与文献[21]中报道的一致,因此可以推断C4和C8属于自陷阱发射机制(STEs).即光激发时,电子被激发至激发态,由于电子-声子之间的强相互作用形成更加稳定的自陷态,自陷激子相对于基态发生扭转,产生宽的荧光发射.
图7 示出了C4和C8在大气环境下存放30 d的PLQY变化,存放的温度为22~25℃,相对湿度为50%~70%,于室内正常光照条件下保存.可以看到C8的稳定性比C4更加优异,其中,C8的PLQY相比初始值下降36%,而C4的荧光7 d之后几乎全部猝灭.这是因为C8具有更长的烷基胺链,能更好地保护[SnBr6]−八面体发光中心,从而提高其稳定性.

Fig.7 Stability curves displayed by the PLQYs of(RNH3)2SnBr4 perovskites
2.3 原位合成的(C8H17NH3)2SnX4的光学性质
类似于铅卤钙钛矿,我们通过调节卤素组成来调控(C8H17NH3)2SnBr4的荧光峰位置.首先采用原位合成法制备了2种(C8H17NH3)2SnX4混卤钙钛矿:(C8H17NH3)2Sn(6Br/I)4(Br/I摩尔比为6∶1)和(C8H17NH3)2Sn(4Br/I)4(Br/I摩尔比为4∶1),为了简便起见,在后续的讨论中分别采用C8-6Br/I和C8-4Br/I表示.
图8 (A)为C8-6Br/I和C8-4Br/I的XRD谱图.与C8一样,二者也具有规律的周期性衍射峰,符合2D钙钛矿的特征.然而谱图的质量较差,出现了一些杂峰,这是由于在反应过程中溴、碘离子快速交换,混卤钙钛矿的晶体结构易发生坍塌.随着I−含量增加,相比于C8-6Br/I,C8-4Br/I的XRD衍射峰向小角度移动,这与Sn-X的晶格膨胀有关,符合文献中报道的规律[22].图8(B1,B2)和(C1,C2)分别为C8-6Br/I和C8-4Br/I的SEM照片,可见两者均具有2D层状形貌.

Fig.8 PXRD patterns of C8⁃6Br/I and C8⁃4Br/I and SEM images of C8⁃6Br/I(B1,B2)and C8⁃4Br/I(C1,C2)
图9 (A1)和(A2)分别为C8、C8-6Br/I和C8-4Br/I(左起1,2,3)在室内光照和365 nm紫外光照射下的光学照片.可以看到,随着Br−逐渐被I−替代,(C8H17NH3)2SnX4表现出从黄色到橙红色的发射.由图9(B1)和(B2)可见,C8-6Br/I和C8-4Br/I的发射峰分别红移至609 nm和620 nm,二者的PLQY值分别为72%和43%.(C8H17NH3)2SnX4的荧光寿命衰减曲线如图9(C1)和(C2)所示,同样采用单指数函数对其进行拟合,C8-6Br/I和C8-4Br/I的荧光寿命分别长达5.10 μs和4.91 μs.综上所述,随着I−含量增加,(C8H17NH3)2SnX4的荧光发射峰发生红移且PLQY逐渐降低.(C8H17NH3)2SnX4具有宽带发射、大的斯托克斯位移和长的荧光衰减寿命,同样属于STEs发射机制.

Fig.9 Photographs under room(A1)and 365 nm UV light(A2),PL emission and PL excitation spectra(B1,B2),time⁃resolved PL decays spectra(C1,C2)of C8⁃4Br/I(B1,C1)and C8⁃6Br/I(B2,C2)
2.4 离子交换反应制备的(C8H17NH3)2SnX4的发光性质
进一步采用离子交换后处理的方法制备了(C8H17NH3)2SnX4钙钛矿,图10为Br/I比不同的(C8H17NH3)2SnX4钙钛矿的荧光发射光谱图.可见,在紫外光激发下,(C8H17NH3)2SnX4从明亮的黄色发光变成微弱的橙红色发光[插图为(C8H17NH3)2SnX4钙钛矿在紫外灯下的光学照片].随着ZnI2用量的增加,发射峰位置逐渐从600 nm红移至622 nm,证明体系中反生了离子交换反应,部分Br−被I−替代.这种变化趋势与文献报道的铅基钙钛矿材料一致[22].

Fig.10 Emission spectra of(C8H17NH3)2SnX4 perovskites with different ZnI2 contents
3 结 论
采用反溶剂法快速合成了发明亮黄色光的二维(C4H9NH3)2SnBr4和(C8H17NH3)2SnBr4钙钛矿,其中(C4H9NH3)2SnBr4钙钛矿具有高达98.5%的PLQY,且通过调控卤素离子比例实现了(C8H17NH3)2SnX4的荧光峰位置调控.相比传统的热注射法,该方法简单易行,室温条件下即可完成,有利于实现2D锡卤钙钛矿的大规模合成.这两种荧光粉材料有望应用于固态照明和显示器件领域.
