自噬参与糖尿病血管病变的研究进展
2021-12-13魏海军张铖杨舒涵郑杨申嘉陵刘勇何延政孙晓磊
魏海军张 铖杨舒涵郑 杨申嘉陵刘 勇何延政孙晓磊*
(1.西南医科大学附属医院血管外科,四川 泸州 646000;2.西南医科大学心血管研究所,四川 泸州 646000;3.四川省卫生健康委员会,成都 610041)
自噬(autophagy)是真核生物特有的一种分解代谢过程,在各种细胞内或细胞外刺激条件下,细胞质中受损的蛋白质、细胞器被双层囊泡膜结构包裹后,与溶酶体融合,使得囊泡内容物降解以供细胞再利用。近年来,关于糖尿病血管损伤的病理生理机制有较多研究,其中高糖环境是导致糖尿病血管损伤的关键因素之一。而自噬作为一种机体的防御机制,在糖尿病血管损伤中发挥的作用不可小视。
1 自噬
1.1 自噬概述及其相关分子
自噬的过程包括:分隔膜、吞噬泡的形成,溶酶体与吞噬泡的融合,内容物的降解与释放等过程。自噬的生理功能主要体现在以下几个方面:(1)胞内蛋白可通过自噬被降解,实现细胞器的更新和转化,从而维持细胞内环境的稳态[1];(2)由自噬产生的降解产物可被机体循环再利用以提供能量[2];(3)自噬作为一种防御机制,能在外界不良刺激和应激条件下产生适应性反应,保护受损的组织或细胞。但是,当自噬被过度激活时,又可作为一种程序性死亡调控程序,调控细胞的死亡[2]。自噬主要以三种不同的形式存在:巨自噬,微自噬和伴侣介导的自噬[3-4],所有这些都促进溶酶体中细胞溶质的蛋白水解降解。巨自噬和微观自噬都能够通过选择性和非选择性机制吞噬大型结构分子。在伴侣蛋白介导的自噬(CMA)中,靶向蛋白与伴侣蛋白(例如Hsc-70)形成复合物,穿过溶酶体膜,该蛋白被溶酶体膜受体溶酶体相关膜蛋白2A(LAMP-2A)识别,从而导致蛋白的变性和退化。
自噬相关基因(autophagy related gene,Atg)负责调控自噬的过程,它的基因片段在进化上相对保守。自噬的调控机制十分复杂,它包含Atg及其所编码的蛋白产物参与自噬的诱导,分隔膜的形成,自噬体的产生、成熟等过程。ULK1复合物(ULK1、FIP200、Atg13)是连接上游刺激信号与下游自噬体形成的桥梁,能接收上游各种信号刺激并做出应答[5],从而诱导自噬产生。而PI3K复合物(Atg6/Beclin-1、Atg14、Vps34/PI3KC3、Vps15)可介导分隔膜的产生[6]。另外,Atg12的结合过程与微管相关蛋白1轻链3(microtubule associated protein 1 light chain 3,LC3)的修饰过程,这两个泛素化过程在自噬体的延伸阶段发挥着重要作用[7]。LC3是由哺乳动物细胞酵母菌Atg8编码产生的一种自噬相关蛋白,被认为是一种自噬标志分子。前体LC3合成之后,被水解切割掉特异片段后,形成LC3I。LC3I又在Atg7和Atg12-Atg5-Atg16 L的作用下,转变为LC3II,始终结合于自噬体膜上,所以LC3II的含量也可以反映自噬体的数量。Beclin-1是由哺乳动物细胞酵母菌Atg6编码产生的一种自噬相关蛋白[8],可介导自噬蛋白定位于吞噬泡,继而调控自噬体的形成与成熟,其表达量的多少亦可反应自噬活性的强弱。
1.2 糖尿病状态下自噬调控的分子机制
目前的研究中,糖尿病通过以下通路影响细胞的自噬:氧化应激反应(oxidative stress)、内质网应激(ER stress)、mTOR依赖性信号通路、AMPK途径等。
2型糖尿病的特征是高血糖和高胰岛素血症,通常伴有糖原和脂质的细胞内蓄积。糖基化和脂肪酸中间体的积累破坏了线粒体和过氧化物酶体的稳定性,导致产生活性氧[9]。并且营养物质的过量也会促进未折叠蛋白和潜在毒性脂质池的形成,从而引起内质网应激[10-11]。自噬通过去除部分去极化的线粒体,将ROS水平维持在一定的水平,另一方面,ROS可以通过抑制mTOR、增加Beclin-1的表达、LC3-I向LC3-II的转化激活自噬[12-15]。此外,ROS可以作为激活JNK-1的信号分子[16],大量的ROS导致线粒体通透性转变孔的开放使线粒体膜电位的破坏,从而导致PINK1/parkin介导的自噬发生[17-19],ROS还可以通过转录水平(在细胞核中)和翻译水平(在细胞质中)调节来诱导自噬过程[20],ROS升高分别激活转录因子HIF-1α、p53、FOXO3、Nrf2,促进BNIP3、NIX、TIGAR、LC3和p62的转录[21]。
内质网应激通过UPR途径和内质网Ca2+转运来诱导自噬[22]。UPR的三个途径:(a)PERK通路——它的激活导致转录因子4(transcription factor 4,ATF4)的翻译[23],(b)IRE1α通路——激活c-Jun-N-terminal激酶(JNK途径[24]和通过移码突变的方式修饰x-box结合蛋白1(x-box binding protein 1,XBP1)的mRNA[25],(c)ATF6通路——调节其他UPR成员包括XBP1[26]。PERK通路通过ATF4诱导ATG5、ATG7、ATG10等自噬相关基因的转录从而影响自噬[27]。PERK通路也促进了聚谷氨酰胺72(polyglutamine 72,PolyQ72)诱导LC3-I向LC3-II的转化过程[28]。在内质网应激的早期阶段,激活的IRE1α通过JUK途径磷酸化参与诱导自噬过程的Bcl-2,从而导致Bcl-2从Beclin-1分离出来[29]。此外,XBP-1—IRE1α途径的另一个参与者,也会促进Beclin-1转录。ATF6通过诱导XBP-1的转录间接参与自噬过程[22]。ER应激下,过度的Ca2+进入细胞质中通过3个不同的机制诱导自噬:(a)刺激依赖CamKK/AMPK途径导致mTOR抑制[30],(b)激活的坏死相关蛋白激酶(death-associated protein kinase,DAPK)通过参与磷酸化Beclin-1[31],(c)激活PKC途径从而导致Bcl2-Beclin-1复合体分离。
高血糖可激活mTOR信号通路减轻细胞自噬。mTOR的机制靶标是经典的营养途径,通过与两种不同的蛋白复合物mTOR复合物1(mTORC1)和mTOR复合物2(mTORC2)结合来调节自噬活性。其中,mTORC1通过直接磷酸化抑制Ulk1复合物的活性,从而抑制自噬相关蛋白LC3B和Beclin-1的表达[32-33]。mTORC1在高血糖情况下被很大程度的激活[34]。通过特异性地敲低相关基因或者使用雷帕霉素(mTOR的抑制剂)抑制mTORC1的表达,可以促进自噬的恢复可以减轻糖尿病对细胞的产生的不利影响[35]。
AMP激活的蛋白激酶(AMPK)是一种营养敏感的激酶,在葡萄糖不足的情况下,AMPK通过Ser317和Ser777的磷酸化直接激活ULK1,从而促进自噬。相反,高葡萄糖下的mTOR或AKT活性增强导致Ser757处的ULK1磷酸化,抑制ULK1活化,从而破坏ULK1和AMPK之间的相互作用,并减少AMPK下游信号通路的自噬诱导作用[32]。AMPK的激活还可以在Beclin-1与BCL-2分离后刺激JNK1形成VPS34蛋白复合物17,并激活自噬[36]。其他研究也报道,AMPK调节FoxO转录因子的表达,从而诱导自噬相关基因的表达[37]。
2 自噬参与糖尿病相关血管病变
2.1 微血管病变
2.1.1 自噬在糖尿病视网膜病变中的作用
糖尿病视网膜病变(diabetic retinopathy,DR)是一种糖尿病微血管并发症,占致失明原因的5%,其组织学特征包括血-视网膜屏障破裂、新生血管形成、毛细血管无灌注、周细胞脱落、内皮细胞减少和纤维血管增生异常。研究表明,毛细血管闭塞和毛细血管通透性的增加将破坏血-视网膜屏障,是产生血管渗漏的主要原因,而血管内皮生长因子(vascular endothelial growth factor,VEGF)既可促进血管渗漏,又可导致新生血管生成,在DR中扮演了重要的角色。近几年的研究中表明,自噬在糖尿病视网膜病变的新生血管的形成以及血管渗漏中起着重要的作用,而自噬可以调节VEGF的分泌从而减轻相应的病变。
高糖环境可以使细胞中的ROS水平增高,导致炎症反应,从而使释放TNF-α、IL-1β、IL-17A、IL-8和IL-6,在这些炎症介质水平的升高的同时,VEGF连同其可溶性受体sIL-2R的表达也相应增加,VEGF的升高促进新生血管的形成使DR进一步加重[38]。Müller细胞是糖尿病视网膜病变中分泌VEGF较多的细胞之一,研究发现高糖水平可以促进Müller细胞中的自噬标记物升高,但是p62/SQTSM1与内质网应激水平也持续升高,其结果导致溶酶体功能失调使得自噬功能障碍而导致细胞的凋亡,VEGF的释放增加[39];通过转染siRNA抑制p62/SQTSM1的表达以及mTOR的抑制剂Rapamycin,激活cathepsin L的活性使溶酶体水解活性增强,导致自噬体降解,使细胞的自噬功能恢复从而抑制VEGF的分泌。与此类似的研究也说明[40],高糖导致的p62/SQTSM1的表达升高,可以被三七皂苷(notoginsenoside R1,NGR1)通过PINK1依赖的线粒体自噬途径抑制,降低Müller细胞中的ROS水平以及炎症反应,从而降低VEGF的表达;NGR1上调了HG诱导的rMC-1细胞和db/db小鼠视网膜中PINK1和Parkin的水平,增加了LC3II/LC3I的比例,并下调了p62/SQSTM1的水平,从而减少细胞中的ROS水平以及炎症反应,降低VEGF的表达。
高血糖除了可以直接影响p62/SQTSM1的表达以外,还可以损伤血-视屏障导致低密度脂蛋白(LDL)从血管中渗出。经过氧化以及糖基化修饰后的低密度脂蛋白(HOG-LDL)在早期的DR中有很强的驱动作用。然而在这个过程中,诱发的自噬会导致细胞出现死亡以及VEGF的分泌,导致新生血管形成从而加重糖尿病视网膜病变。研究发现Müller细胞经过HOG-LDL刺激后使AMPK激活导致自噬相关蛋白ATG-5、Beclin-1、LC3II/LC3I蛋白水平明显升高,与此同时凋亡相关蛋白caspase-3的表达也进一步升高说明自噬的激活会导致细胞凋亡[41]。他们的研究中使用苄基四羟基喹啉(benzyltetrahydroxyquinoline,berberine)可以抑制AMPK的激活减轻细胞中HOG-LDL导致的自噬性细胞死亡、炎症反应以及血管新生等。近几年的一些研究通过高糖培养视网膜内皮细胞(retinal endothelial cells,RECs)来建立糖尿病视网膜的血管新生病变的体外模型来进一步探寻DR的内在机制。Mao等[42]研究提示去乙酰化酶3(Sirtuin 3,Sir3)可以抑制HRECs中迁移相关因子MMP-2、MMP-9、血管新生因子VEGF、HIF-1a、和IGF-1的表达,而且研究中发现过表达Sir3可以促进LC3I以及LC3II的表达以促进细胞自噬,并且细胞自噬后期可以降低VEGF、HIF-1α的表达,故Sir3可能通过自噬来抑制HRECs的血管新生。Mao等[43]进一步研究发现在体内实验中和体外实验中发现miR-204-5p可以促进糖尿病大鼠视网膜的血管新生,其中LC3II以及LC3I/LC3II的表达降低,说明通过降低miR-204-5p的表达可以促进LC3II的表达从而减低VEGF的分泌。
综上所述,自噬在糖尿病视网膜病变中在多个细胞中起着调节作用,目前大部分的研究肯定了自噬在DR中的保护作用,但是在某些情况,过度自噬也将会导致DR进一步恶化[41],这两种作用的相互联系尚需进一步研究阐明。
2.1.2 自噬在糖尿病肾病中的作用
糖尿病肾病(diabetic nephropathy,DN)作为糖尿病最严重的并发症之一,目前已成为继肾小球肾炎后终末期肾病的第2位原因,DN的一个重要临床特征是尿蛋白。在蛋白尿的糖尿病患者中发现肾小球滤过屏障的结构改变,包括肾小球内皮细胞损伤[44-45]。近来有越来越多的证据表明,肾小球血管周围的足细胞自噬水平对糖尿病肾病发展有重要的作用。在不同类型的细胞中,自噬发挥的作用也有所不同:
(1)足细胞 足细胞损伤是DN中关键因子,可以作为DN演变的临床预测指标。在高糖的反复刺激下,维持细胞内稳态对于足细胞的命运极为重要[46]。近几年的研究中发现,自噬可以维持足细胞的稳态,从而改善糖尿病肾病。研究发现,相当一部分肾保护药物是通过mTOR/AMPK途径促进自噬相关蛋白如LC3II、Beclin-1等的表达,增强自噬形成,从而对足细胞产生保护作用[47-53]。Liu等[54]研究发现,在高血糖下,足细胞中β-arrestin-1和β-arrestin-2的表达水平增高,而且β-arrestin-1和β-arrestin-2可以通过抑制ATG12-ATG5的结合来抑制足细胞自噬,导致肾的损伤,这可能成为治疗糖尿病肾病新的靶点。
既往的研究显示,在糖尿病肾病的动物模型中发现了上皮-间充质转换(epithelial-mesenchymal transition,EMT)的病理现象。EMT是一个动态的发展过程,其特征在于上皮细胞获得间充质细胞(如成肌纤维细胞)的特征,这会导致上皮细胞标记物(如E-钙黏着蛋白)表达降低,间充质标记物(如α-SMA和纤连蛋白)的表达增强,从而破坏肾小管基底膜以及增强足细胞的迁移[55]。Jin等[56]发现高糖培养下,足细胞中STIM1激活TRPC6和Orai1通道,使钙离子通道打开,导致p62表达增高和LC3II/LC3I降低从而抑制自噬,最终加速了EMT的发生。Jin等[57]在进一步研究后发现,HMGB1通过激活AKT/mTOR通路抑制LC3II的表达和促进p62的升高,从而导致足细胞的EMT的增强。这些研究说明,自噬抑制足细胞的EMT,从而抑制糖尿病肾病的进展。
(2)肾小球内皮细胞 Lenoir等[58]发现内皮细胞自噬与糖尿病肾病之间相关的直接证据。他们研究显示,内皮细胞特异性自噬缺陷小鼠会出现严重的肾小球损伤,表明内皮细胞自噬对糖尿病所引起的应激起着重要的保护作用;并且内皮细胞特异性自噬缺陷小鼠模型可提供足细胞和近端肾小管细胞以外的其他肾细胞中糖尿病肾病自噬的重要信息。晚期糖基化终末产物(AGEs)和AGEs受体(RAGE)被证明在糖尿病性肾病中起重要作用。Hou等[59]研究表明丹酚酸A(salvianolic acid A,SalA)通过AGE-RAGE-Nox4轴减弱了AGEs诱导的ROS,从而促进Atg5、Atg7、Atg12、LC3II/LC3I的表达以及抑制p62的表达,从而恢复高糖导致的内皮细胞自噬紊乱,抑制DN的进程。Lim等[60]的研究表明,西那卡塞(Cinacalcet)通过CaMKKβ-LKB1-AMPK轴促进肾小球内皮细胞中LC3II/LC3I、Beclin-1的表达,从而减轻高糖对肾小球的损伤。这些研究证明,肾小球内皮细胞中自噬的激活对肾脏功能起着保护作用。
2.2 外周血管病变
2.2.1 自噬在糖尿病动脉粥样硬化性病变中的作用
动脉粥样硬化是导致心脑血管疾病以及下肢动脉硬化的主要原因,它是以脂质蓄积和炎症细胞浸润为特征的慢性炎症性过程。在糖尿病导致的动脉硬化疾病中,自噬起着调节作用。Tian等[61]的研究发现,高糖激活mTOR信号通路抑制LC3II/LC3I以及Beclin-1的表达以及增加SQSTM1/p62堆积抑制自噬,导致内质网应激相关蛋白如p-JNK、CHOP和Caspase-12表达升高,诱导凋亡,从而促进链脲菌素诱导的糖尿病ApoE-/-小鼠动脉粥样硬化。Tian等[62]的研究发现,糖尿病状态下,糖化高密度脂蛋白引起巨噬细胞的凋亡并伴随内质网应激活化—包括核转位激活转录因子6、蛋白激酶样ER激酶(PERK)的磷酸化和真核生物翻译起始因子2以及C/EPB同源蛋白的上调,通过加入自噬诱导剂雷帕霉素(Rapamycin,RAPA)可以抑制上述过程,从而抑制巨噬细胞的凋亡;而加入自噬的抑制剂3-MA或抑制Beclin-1表达可以促进这一过程,导致巨噬细胞的凋亡,从而促进糖尿病动脉粥样硬化性疾病的进展。Lazaro等[63]的实验也证明核样因子2(nuclear factor(erythroid-derived 2)-like 2)可以通过促进自噬相关蛋白Beclin-1、LC3II/LC3I的表达升高,SQSTM1表达的降低来抑制血管壁的炎症相关水平,使糖尿病导致的动脉粥样硬化向好的方向发展。
动脉粥样硬化的始发事件是血管内皮细胞的损伤[8]。高血糖可导致内皮细胞内的活性氧水平上升,从而导致炎症,内皮细胞的损伤最终导致细胞凋亡。Xie等[64]的研究中发现,AGEs可以使血管内皮细胞中的活性氧水平升高,作为一种防御机制,机体内的自噬水平相应的增加,而且自噬相关的抑制剂3-MA加重了AGEs对内皮细胞的损伤。Fetterman等[65]研究发现在内皮细胞中,自噬可以激活一氧化氮合酶(eNOS);通过从糖尿病病人以及正常人中收集内皮细胞分析发现,相比于正常人来讲,糖尿病病人内皮细胞的p62水平明显升高,标志着自噬通量降低。虽然糖尿病组与正常组的LC3以及Beclin-1水平相似,但是在糖尿病组中的自噬终末期的标志物溶酶体蛋白Lamp2a的水平明显升高,通过抑制终末期的自噬,使自噬长期处于活跃水平导致eNOS激活,从而减轻内皮细胞的损伤。Bai等[66]的研究中提出高血糖导致动脉硬化的新机制,他们的研究指出内皮细胞中的小窝蛋白1(caveolin-1,CAV1)具有介导脂质大分子物质胞吞的作用,它有两种类型的LC3相互作用区(LC3-interacting region,LIR):CAV1支架结构域(the CAV1 scaffolding domain,CSD)和膜内结构域(intramembrane domain,IMD)。CAV1可 以 与CAVIN1-LC3B结合并形成复合物。由于小窝相关蛋白1(caveolae associated protein 1,CAVIN1)没有LC3结合片段,而CAV1具有超过2个LIR,所以CAV1作为CAVIN1和LC3B之间的中间连接体可以介导自噬抑制。当CAV1作为结构蛋白存在于膜小窝或小窝中以介导LDL转胞作用时,IMD中的LIR被掩埋,CAV1仅对通过CSD的自噬发挥抑制作用。当CAV1存在于细胞质中时,CAV1通过IMD可能有机会与LC3B结合并介导CAV1的自噬降解。在正常的葡萄糖环境下,CAV1在细胞质中的自噬降解和利用CAV1形成膜小泡保持了一定的稳态,并且在内皮细胞膜中小泡的形成相对较少,因此内吞小窝对LDL的胞吞作用也维持在相对较低的水平。通过抑制AMPK-mTOR-PIK3C3途径,高葡萄糖抑制了CAV1-CAVIN1-LC3B介导的CAV1自噬降解。结果,更多的CAV1积累在细胞质中,在细胞膜中形成更多的小孔并促进LDL跨内皮细胞的胞吞作用,从而促进动脉粥样硬化脂质在内皮下的滞留,这个机制或许可为糖尿病导致动脉粥样硬化提供新的治疗方案。
与此同时,其他的一些研究也发现自噬在内皮功能保护中的不利作用。Cai等[67]的研究发现,与正常组比较,高糖组自噬相关蛋白Atg7、LC3II/LC3I的水平明显升高,而且这个过程中凋亡相关蛋白Bax、BCL-2蛋白水平也明显升高,说明高糖可能导致内皮细胞的自噬性死亡,而GLP-1高血糖素样肽-1(glucagon-like peptide-1,GLP-1)通过GLP-1RERK1/2依赖性通路,恢复HDAC6活性,抑制高糖导致内皮细胞功能障碍和过度自噬性死亡,提示自噬在内皮细胞中有害的一面。除此之外,Qiu等[68]发现,高糖降低血管平滑肌细胞内硫化氢的水平,激活AMPK/mTOR通路导致LC3II/LC3I、Beclin-1的水平升高以及P62降低,而这个自噬增强的过程可以导致平滑肌细胞的功能紊乱,也提示了自噬在糖尿病动脉粥样硬化过程中的不利作用。
2.2.2 自噬在糖尿病血栓形成以及瘤样扩张状态中的作用
大约65%的糖尿病患者死于血栓性事件,包括心脏病发作和中风[69]。其中大约40%~60%的糖尿病病人对抗血栓药物阿司匹林耐受[70],进一步的研究发现糖尿病病人血液中血小板被显著激活[71]。Lee等[71]研究发现,高糖可以导致血小板内的ROS水平升高,pp53的激活,线粒体功能紊乱,导致血小板的凋亡从而形成血栓;而激活线粒体自噬可以维持线粒体功能抑,制血小板的功能紊乱,从而抑制血栓的形成。Paul等[72]通过饥饿的方式抑制血小板的聚集,自噬相关的因子Atg3、Atg5、Atg7、Atg12、Atg16、Beclin-1、LC3II/I明显上升,加入自噬抑制剂3-MA可以恢复血小板的聚集,进一步证实了自噬在血小板的聚集中的有利的作用。
主动脉中膜平滑肌细胞的减少导致中膜薄弱可以导致动脉瘤的形成,糖尿病状态下,腹主动脉瘤与胸主动脉瘤的发病率明显降低[73]。Zheng等[74]发现,腹主动脉瘤患者组织中自噬相关基因转录因子TG4b、Atg6/Beclin-1,Bnip3和Vps34等表达升高,伴随整合素/CD44的激活和p38/MAPK信号通路的增强,同时,LC3蛋白表达水平且LC3II/I值也相应增高。另外,Giusti等[75]通过微阵列分析技术对腹主动脉瘤患者和正常人的外周血进行基因表达谱差异分析,发现自噬相关基因Atg5表达上调。因此,腹主动脉瘤患者中自噬水平的升高很可能与机体自身的防御抵抗机制密切相关。近来又有研究发现,RAPA可极大地延缓腹主动脉瘤的发病进程,但RAPA同时也是一种炎症抑制剂,故不能完全确定RAPA是否通过激活自噬而减轻腹主动脉瘤的进展[76]。这些研究都证实自噬和高血糖可能参与腹主动脉瘤的病理过程,但目前尚没有更多研究阐明自噬、糖尿病以及动脉瘤之间相互关系。
上述研究结果显示,虽然自噬相关基因在糖尿病外周血管病变中上调,但自噬在糖尿病外周血管病变发病机制中的具体机制仍未阐明。体外实验中,许多糖尿病外周血管病变相关因子可以激活自噬而发挥保护性作用,且此保护作用是通过减弱氧化应激和炎症信号转导而起效;体内实验中,越来越多的证据表明自噬在动脉粥样硬化中起保护作用,且可维持动脉粥样硬化斑块的稳定性。其中,RAPA减轻糖尿病外周血管病变的机制尚不清楚,但其为激活自噬治疗糖尿病外周血管病变提供了新的方向。
3 结语与展望
作为机体的防御机制和蛋白质降解的重要途径,自噬在糖尿病血管病变中的作用正在渐渐被探索、挖掘。本文综述了自噬分别在糖尿病视网膜病变、糖尿病肾病、糖尿病外周血管病变、糖尿病心脑血管病变中的作用,并对自噬参与糖尿病血管病变的分子生物学机制进行了深入探讨(图1)。就目前的研究趋势来看,通过使用自噬激活剂和抑制剂来调控自噬的水平已经成为了学者们的惯用思路,当然,过表达特定自噬相关基因或使其被敲除也是目前常用的研究方法。但是大多数研究都是从细胞水平出发探究自噬在相关病理状态下的作用,也使得其后续的功能检测方法受限,因而建议从整体水平更加全面地探究自噬在糖尿病血管病变中的作用。并且,自噬在某些糖尿病血管病变中的病理生理作用仍未阐述清楚,部分结论仍有争议,所以未来仍需深入研究。糖尿病血管病变的病理生理学机制十分复杂,因此找寻其发病机制中的关键因子或重要的信号通路就显得尤为重要,通过自噬筛选的相应靶点可以作为治疗糖尿病血管病变的新途径。虽然目前自噬在糖尿病血管病变中的发生机制仍未阐明,但相信随着研究的不断深入,靶向自噬的机制研究一定会为糖尿病血管病变的治疗开创新局面。
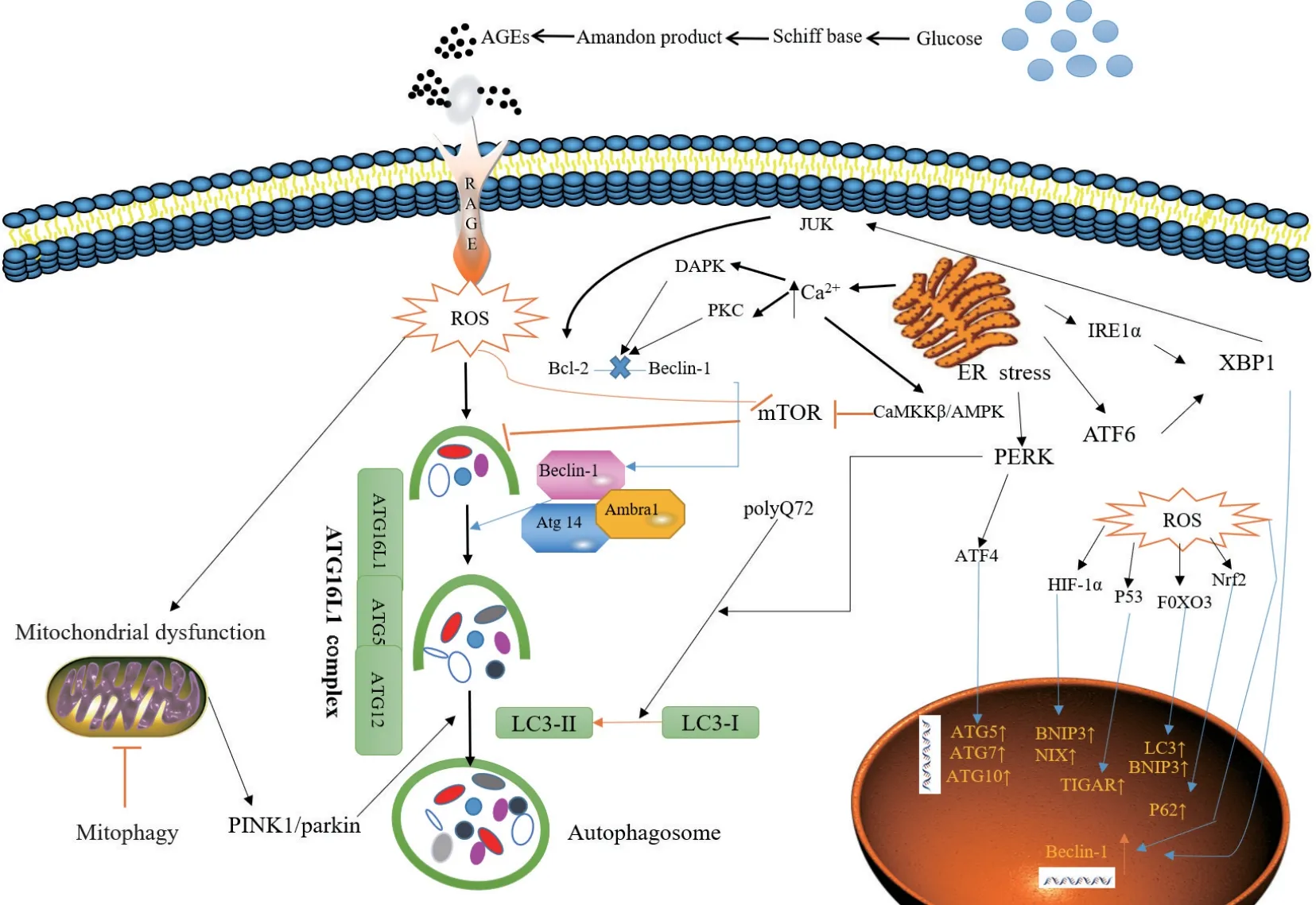
图1 糖尿病状态下自噬调节的信号通路Figure 1 Signal pathways regulating autophagy in diabetes
