两个含氮、氧杂原子有机配体的铀酰配合物溶剂热自组装合成、结构及其性质
2021-12-09蒋伍玖李骜典唐铭豪南小龙谭延亮谭宇星
蒋伍玖 李骜典 唐铭豪 南小龙 谭延亮 谭宇星*,
(1衡阳师范学院化学与材料科学学院,功能金属有机材料湖南省普通高等学校重点实验室,功能金属有机化合物湖南省重点实验室,湘江上游重金属污染监测与治理湖南省工程研究中心,衡阳 421008)
(2湖南省核工业地质局306大队,衡阳 421008)
(3衡阳师范学院物理与电子工程学院,铀矿勘查技术湖南省工程研究中心,衡阳 421002)
0 引言
铀是一种放射性金属元素,它是自然界中存在的最重要的核燃料,是核能发展过程中备受关注的一个元素[1-3]。在核燃料循环首端,铀的开采与纯化过程中不可避免地会出现铀的释放;在核燃料循环末端,放射性废物中也会含有大量未反应的铀[4]。目前,世界上许多国家都在加紧核废料的处置研究,期望可以利用化学方法进行核废料的处理与再利用。因此,研究铀的配位化学,了解铀元素的成键特性,探讨新颖铀酰配合物的结构与性质,可以为解决核能发展所面临的核废料安全储存以及核泄漏造成的放射性污染等问题提供实验积累和新的思路。
铀的电子层构型为[Rn]5f36d17s2,其5f轨道的中子对外层电子具有屏蔽效应,使得铀具有多变的氧化态,其中+6价最为稳定,高氧化态的中心离子正电性高,离子半径大,对配体的吸引力强,能够吸引更多的配体从而形成高配位数的配合单元[5-9]。因此,我们设计了2组有机配体(L1=N′,N‴-((2E,3E)-butane-2,3-diylidene)bis(4-hydroxybenzohydrazide),L2=2,4,5-三氟-3-甲氧基苯甲酸,Phen=菲咯啉)与醋酸双氧铀的自组装反应,通过“一锅法”得到了2个未见报道的铀酰配合物,并且研究了配合物的热稳定性、光谱性质以及量子化学,为铀酰配合物在核废料处理、催化、矿物学和能源学等方面的研究提供一定的理论基础。
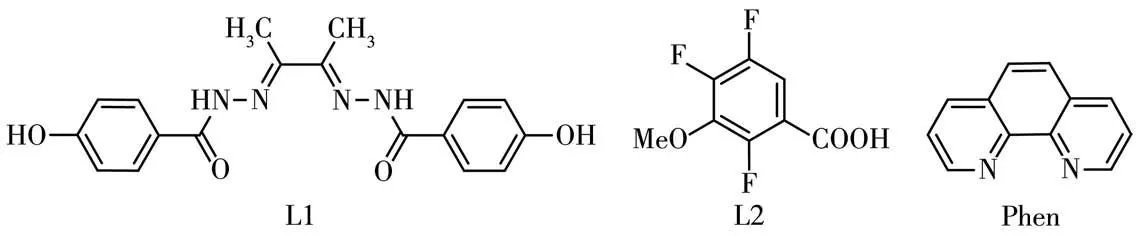
图1 配体的结构Fig.1 Structure of the ligands
1 实验部分
1.1 仪器和试剂
IR用日本岛津Prestige-21红外光谱仪(4 000~400 cm-1,KBr压片)测定。元素分析用PE-2400(Ⅱ)元素分析仪测定。晶体结构用Bruker SMART APEXⅡCCD单晶衍射仪测定。UV-Vis光谱用日本岛津公司UV-2550型紫外可见光谱仪测定。荧光光谱用日本日立F-7000荧光光谱仪测定。热重(TG)和微商热重(DTG)曲线用德国NETZSCH TG 209 F3热重分析仪测定。熔点用北京泰克X-4双目体视显微熔点测定仪测定(温度计未经校正)。
实验中所用的试剂均为分析纯试剂,未经进一步纯化而直接使用。
1.2 配合物的合成
配合物的合成路线如图2所示。
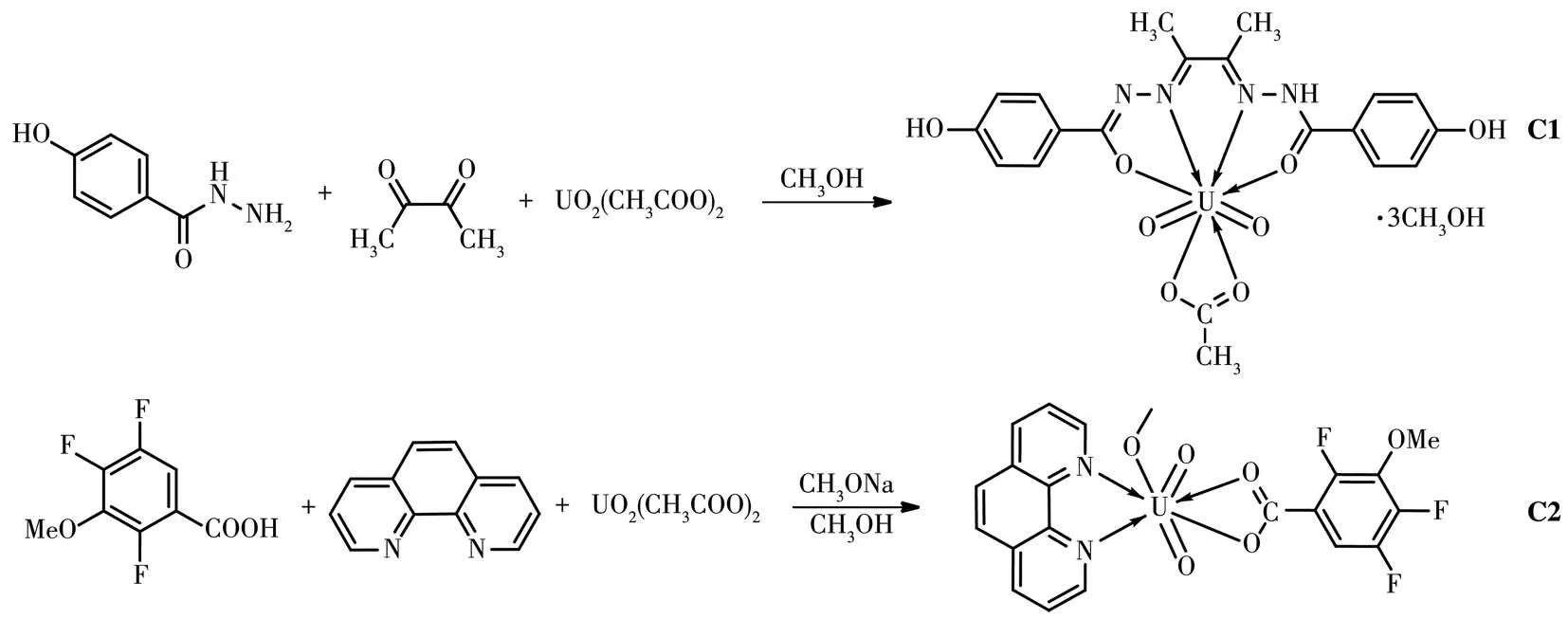
图2 配合物的合成线路Fig.2 Synthesis routes of the complexes
[UO2(L1)(CH3COO)]·3CH3OH(C1):在 20 mL 的水热反应釜中加入1 mmol对羟基苯甲酰肼、0.5 mmol 2,3-丁二酮、0.5 mmol醋酸双氧铀和10 mL甲醇,将体系混合均匀后密封,置于120℃的烘箱中反应24 h,再通过程序降温(5℃·h-1)逐渐恢复至室温。过滤,用甲醇淋洗,得到配合物C1的黄色块状晶体,产率 56%。m.p.65~67 ℃(dec)。元素分析(C23H32N4O11U)实测值(计算值,%):C,35.52(35.48);H,4.16(4.14);N,7.16(7.20)。IR(KBr,cm-1):3 566(s),3 404(m),3 154(m),3 011(w),2 974(w),2 814(w),1 599(s),1 549(s),1 504(s),1 472(s),1 449(s),1 377(s),1 329(s),1 292(s),1 242(s),1 192(s),1 171(s),1 123(w),1 096(m),1 061(m),993(m),912(s),851(m),827(m),758(m),687(m),629(m),546(w),509(w),444(w),436(w)。
[UO2(L2)(Phen)(CH3O)](C2):在20 mL的水热反应釜中加入0.5 mmol L2、0.5 mmol Phen、0.5 mmol甲醇钠、0.5 mmol醋酸双氧铀和10 mL甲醇,将体系混合均匀后密封,置于120℃的烘箱中反应24 h,再通过程序降温(5℃·h-1)逐渐恢复至室温。过滤,用甲醇淋洗,得到配合物C2的黄色块状晶体,产率59%。m.p.220~222℃。元素分析(C21H15F3N2O6U)实测值(计算值,%):C,36.84(36.75);H,2.19(2.20);N,4.08(4.08)。 IR(KBr,cm-1):3 057(w),2 914(w),2 876(w),2 808(w),1 624(w),1 587(w),1 574(w),1 520(s),1 456(s),1 422(s),1 348(m),1 150(m),1 107(m),1 067(s),1 003(w),891(s),860(s),822(m),789(m),725(s),675(s),640(m),436(s),420(m),401(m)。
1.3 晶体结构测定
采用经石墨单色化的MoKα射线(λ=0.071 073 nm),以φ-ω扫描方式收集衍射数据。全部数据经Lp因子和多重扫描吸收校正。晶体结构由直接法解出,全部非氢原子坐标在差值Fourier合成中陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对氢原子和非氢原子分别采用各向同性和各向异性热参数进行全矩阵最小二乘法修正,全部结构分析计算工作采用SHELX-2014程序系统完成。
CCDC:2077418,C1;2077419,C2。
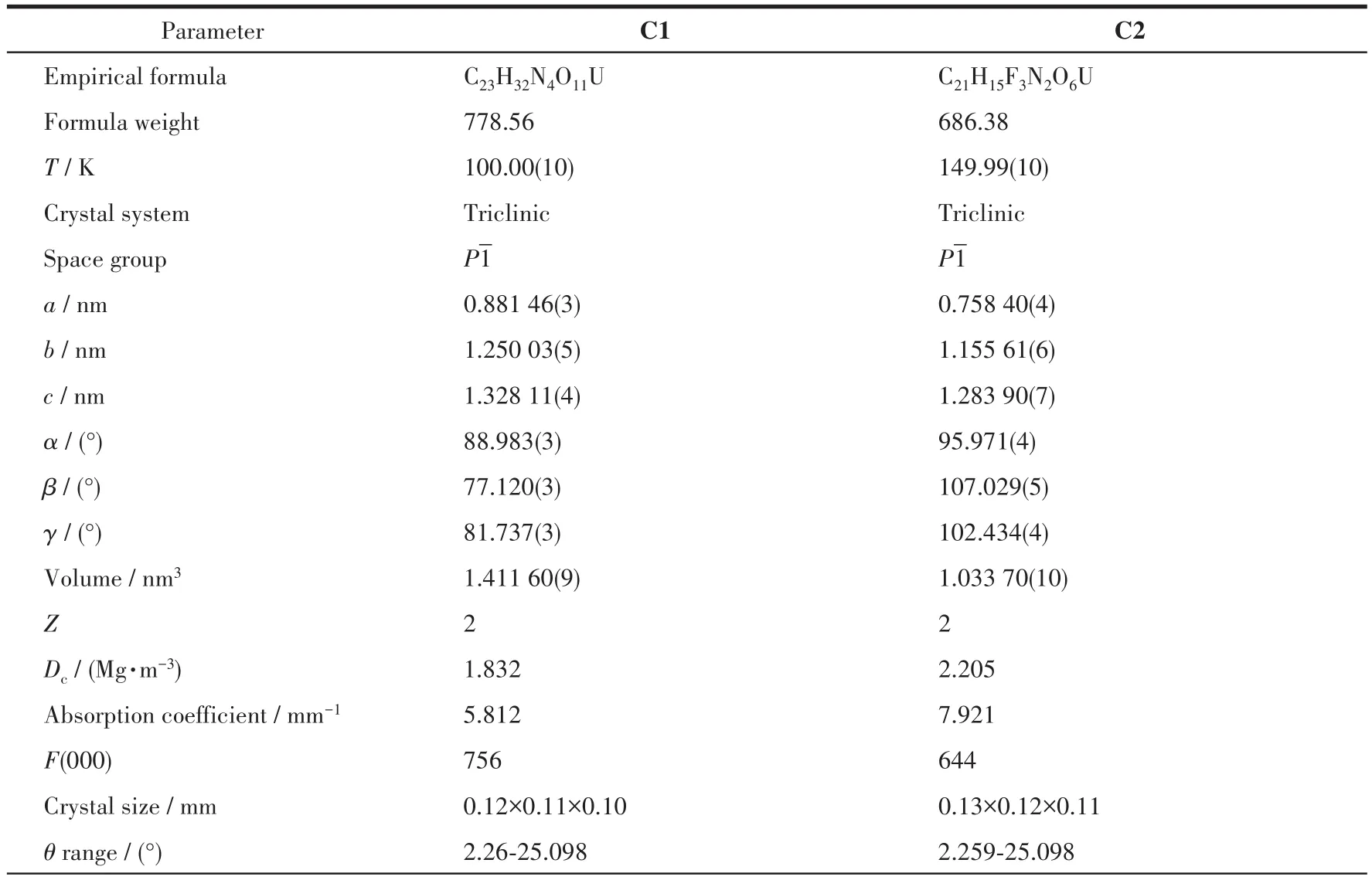
表1 配合物C1和C2的晶体学数据Table 1 Crystallographic data of complexes C1 and C2

续表1
1.4 热稳定性测定
采用NETZSCH TG 209 F3热重分析仪,在空气氛、加热速度20 ℃·min-1、气体流速20 mL·min-1的条件下,对配合物在30~800℃范围内进行了TG测试。
1.5 光谱性质研究
在室温条件下230~600 nm波长范围内测定配体(L1、L2、Phen)和配合物(C1、C2)的 UV-Vis光谱。溶液浓度为0.1 mmol·L-1,溶剂为甲醇,参比为甲醇。配体L1参考文献方法[10]合成。
配体(L1、L2、Phen)和配合物(C1、C2)的荧光光谱由日立F-7000分光光度计测定。溶液浓度为0.1 mmol·L-1,溶剂为甲醇。发射和激发的狭缝扫描宽度均为5.0 nm。
1.6 量子化学研究
根据配合物晶体结构的原子坐标,运用Gaussian09W程序和B3ylp/mwb,计算得到配合物分子的前沿分子轨道能量。
2 结果与讨论
2.1 合 成
采用溶剂热合成法,在一定温度与自生压力下,将反应物进行自组装形成最终产物。溶剂热法具有普通合成方法无法比拟的优点[11-13],例如,(1)在高温条件下,反应釜内溶剂气化产生自生压力,使得常温下一些比较难溶的配体得以溶解,同时高温条件使得溶剂黏度下降,有利于物质之间的传递;(2)溶剂反应条件简单易行、快速高效,条件容易控制,重现性也较好;(3)在该条件下,通过有机配体的自组装,可以得到具有意料之外的结构的新颖化合物。在配合物C1中,我们可以看到本是对称结构的配体分子,但是与UO22+发生配位时,两边羟基却采取了不同的配位模式,O2与UO22+之间采用的是共价键,而O3与UO22+之间是配位键。在配合物C2的合成中,添加甲醇钠的目的起初是为了使得2,4,5-三氟-3-甲氧基苯甲酸(L2)上的氢离子更容易离去,便于参与UO22+的配位,但是意想不到的是甲醇钠还提供了一分子的甲氧基参与配位。由此可见,有机配体的自组装反应确实可以得到很多结构新颖的化合物分子。
2.2 晶体结构
配合物C1、C2的主要键长和键角数据列于表2,分子结构见图3、4。在配合物C1中,六价铀离子处于八配位的十二面体环境中,UO22+与来自双酰腙上的2个氮原子N2、N3和2个氧原子O2、O3以及醋酸上的2个氧原子O10、O11形成六角双锥构型,这6个原子构成了六角双锥的赤道平面,UO22+上的2个氧原子构成双锥的轴。铀与赤道平面上的氧原子形成的键长分别为U1—O2 0.242 1(4)nm,U1—O3 0.239 0(4)nm,U1—O10 0.249 4(4)nm,U1—O11 0.248 4(4)nm,铀与氮形成的键长分别为U1—N2 0.262 7(5)nm,U1—N3 0.258 3(5)nm,均与文献[14-16]中报道的铀酰配合物中的U—O、U—N键长相符合。2个铀酰氧原子从轴向与铀成键,其U=O键长分别为 0.176 4(4)、0.177 9(4)nm,两者略有差异,键角O=U=O为176.0(2)°,可以近似地认为铀与2个铀酰根氧原子在一条直线上。此外,配合物C1中存在丰富的O—H…O氢键作用,使得配合物构成一维无限链状结构,如图5和表3所示。
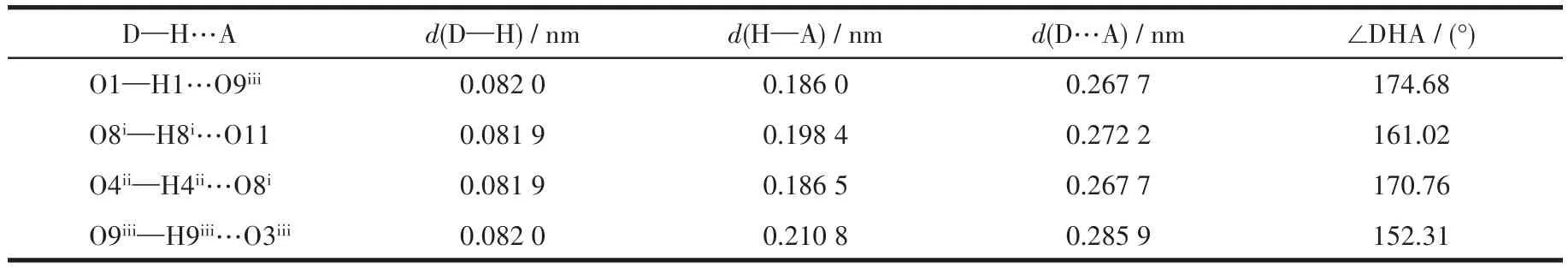
表3 配合物C1的氢键Table 3 Hydrogen bond of complex C1

图3 配合物C1的椭球率50%分子结构图Fig.3 Molecular structure of complex C1 with 50% probability ellipsoids
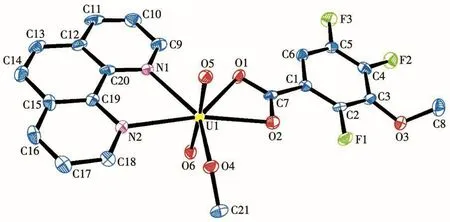
图4 配合物C2的椭球率50%分子结构图Fig.4 Molecular structure of complex C2 with 50% probability ellipsoids

图5 配合物C1的一维链状结构Fig.5 One-dimensional chain structure of complex C1
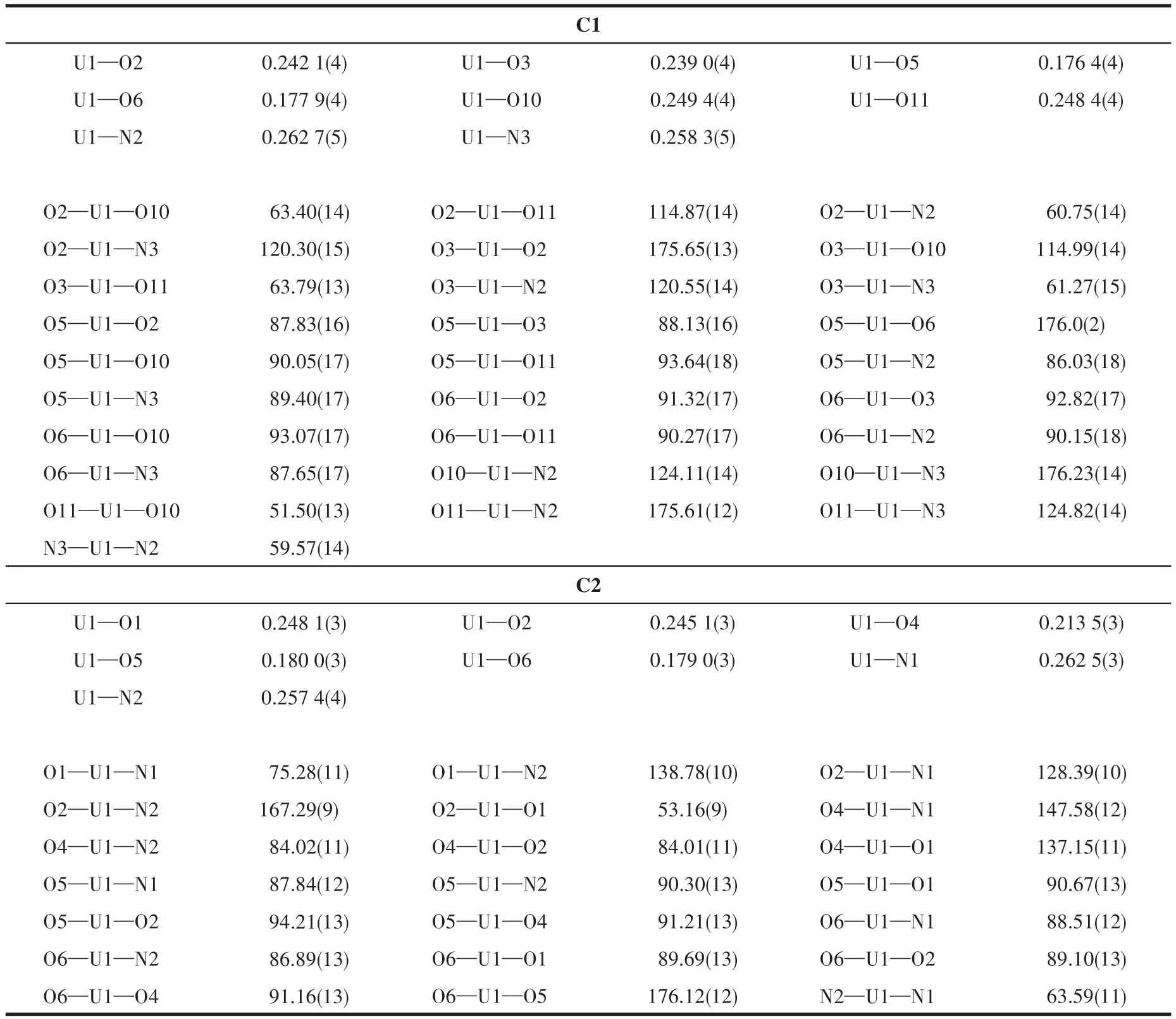
表2 配合物C1和C2的部分键长和键角Table 2 Selected bond lengths(nm)and bond angles(°)of complexes C1 and C2
在配合物C2中,Phen作为一个双齿配体,分子上的2个氮原子参与配位,并且甲醇钠也提供了一分子的甲氧基参与配位,使得六价铀离子处于七配位的十面体的配位环境中。UO22+与来自L2上的2个氧原子O1、O2和Phen上的2个氮原子N1、N2以及甲氧基上的1个氧原子O4形成五角双锥构型。铀酰根的氧原子作为轴,O1、O2、O4、N1、N2构成了五角双锥的平面。在轴向方向,2个铀酰根氧原子从轴的两侧与铀成键,其U=O键长分别为0.179 0(3)、0.180 0(3)nm,键 角 O=U=O 为176.0(2)°,与配合物C1中的O=U=O键角相等,C2的其他化学键也均与文献[17-18]报道的铀酰配合物一致。此外,在配合物C2中,相邻分子之间存在π-π堆积作用(图6),环心间距为0.349 4 nm,夹角为76.943°,与同类配合物之间的π-π堆积作用强度差异不大[19-20]。

图6 配合物C2的π-π堆积Fig.6 π-π stacking of complex C2
2.3 谱学研究
在配合物C1、C2的红外谱图中(图7、8),分别位于 912、891 cm-1处的强峰和 827、822cm-1处的弱峰归属为铀酰根离子的对称伸缩振动和不对称伸缩振动峰,是铀酰类配合物的特征峰,这与文献[21-22]报道的吸收峰位置一致。配合物C1中位于3 566和3 404 cm-1处的峰分别归属为配合物上酚羟基伸缩振动吸收峰和仲胺上的N—H伸缩振动吸收峰。配合物C1中位于3 154和3 011 cm-1处的峰、C2中位于3 057 cm-1处的峰均归属为配合物芳环上的C—H伸缩振动吸收峰,而C1和C2分别位于2 974、2 814 cm-1处和2 914、2 876、2 808 cm-1处的峰归属为饱和C—H伸缩振动吸收峰。配合物C1、C2中的—COO的不对称伸缩振动和对称伸缩振动吸收峰分别位于1 599、1 449 cm-1和1 587、1 456 cm-1处,二者的差值分别为150、131 cm-1,这说明2个配合物中的羧酸根是以双齿形式与金属离子配位,这与X射线单晶衍射所表征的结构一致。
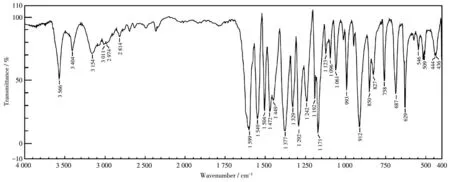
图7 配合物C1的红外谱图Fig.7 IR spectra of complex C1
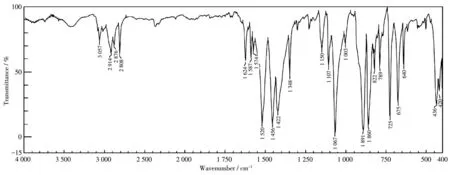
图8 配合物C2的红外谱图Fig.8 IR spectra of complex C2
2.4 热稳定性研究
如图9、10所示,随温度的升高,配合物C1、C2均发生了明显的失重过程。从失重曲线来看,配合物C1在低于100℃时就有明显的失重现象,直到290℃时曲线出现一小段平台,这一阶段对应于配合物C1失去3个游离的甲醇分子,290℃之后配合物C1的骨架结构才开始出现分解;而配合物C2在220℃左右出现失重,晶体结构解析表明该分子中不存在溶剂分子,推测此处出现的失重为配合物骨架结构分解。配合物C1、C2的失重曲线最终稳定在约34.8%(C1)和39.1%(C2),残余物与UO2的计算含量34.6%(C1)及39.3%(C2)吻合,说明2个配合物在升高温度的过程中均失去所有参与配位的配体。上述热分析结果表明配合物C1中含有溶剂分子,但是其骨架结构比配合物C2稳定。
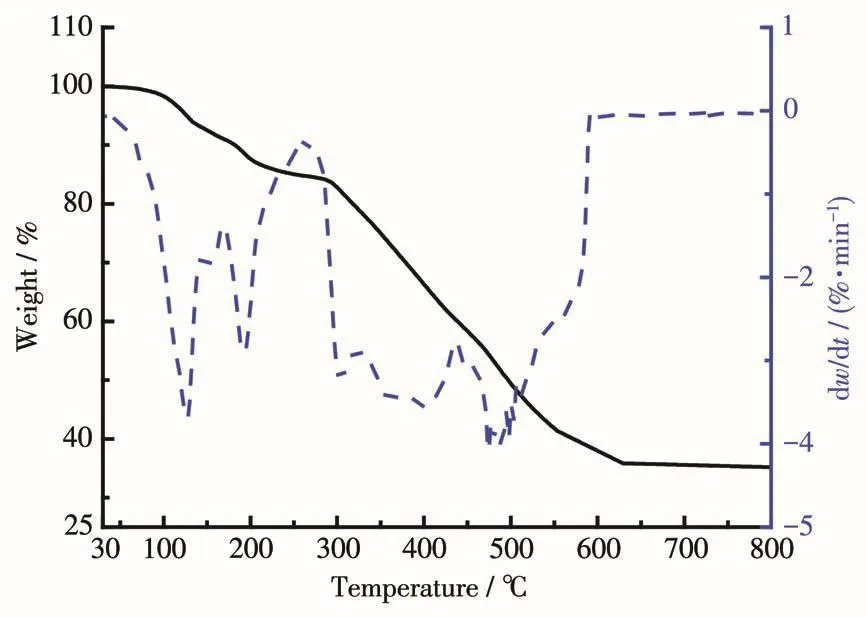
图9 配合物C1的TG和DTG曲线Fig.9 TG and DTG curves of complex C1
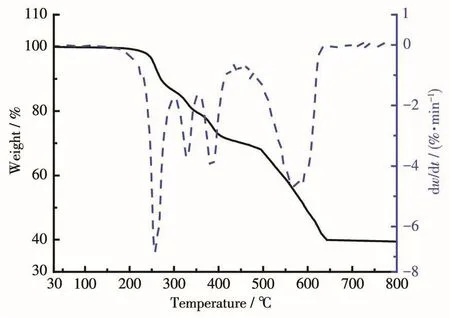
图10 配合物C2的TG和DTG曲线Fig.10 TG and DTG curves of complex C2
2.5 紫外可见光谱
如图11所示,配体L1在254 nm处有一明显吸收带,对应于配体的π-π*跃迁,配合物C1有2个吸收带:在284 nm处有一个弱的吸收带,在433 nm处有一个强的吸收带。在284 nm处的吸收带可以认为是配体吸收带红移导致,仍是配合物中配体部分的π-π*跃迁[23],在433 nm处的强吸收带可能是由于配体与铀原子发生配位作用,形成一个共轭环,引起配体C=N键的极化并影响了相关共轭分子轨道的能级所致,而在配合物C2中未发现这一吸收带,推测配合物C2中的共轭效应没有C1强,X射线单晶衍射测得的结果也证实了这一点。配体L2和Phen与配合物C2的吸收带非常相似,仅有一个吸收带,位置也相差不大,推测配合物C2在262 nm处的吸收带归属为配体部分的π-π*跃迁。
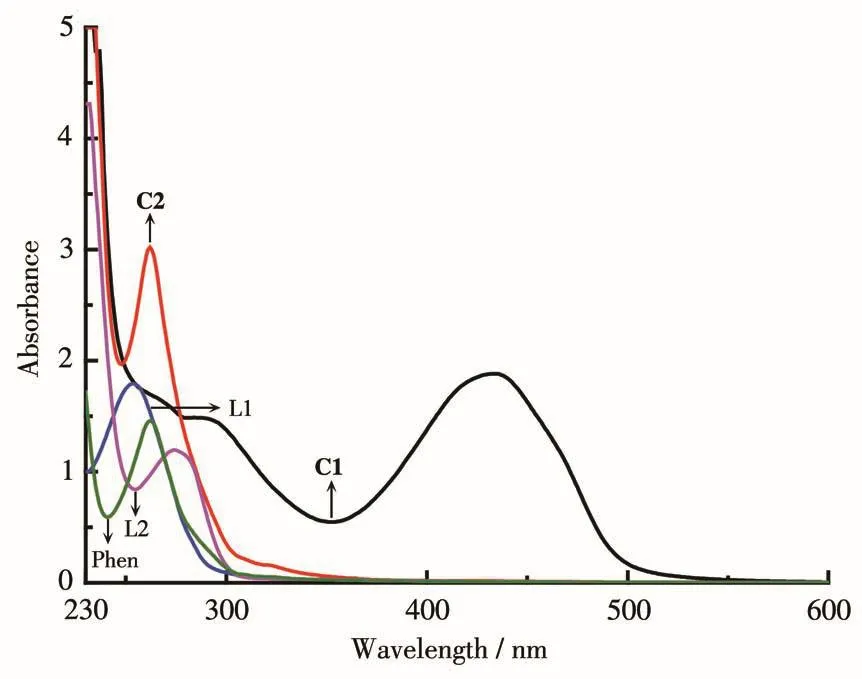
图11 配体和配合物的UV-Vis吸收谱图Fig.11 UV-Vis absorption spectra of the complexes and the ligands
2.6 荧光光谱
通过3D荧光扫描得到配合物C1和C2的激发波长分别为410和310 nm,在此激发波长下,配合物C1、C2分别在466和382 nm处产生了明显的发射峰,如图12所示。配合物C1、C2能够产生荧光可以归因于从配体到金属离子的电荷转移(LMCT),这也进一步表明了醋酸双氧铀与配体配位形成配合物,而且从C1、C2的荧光强度来看,显然配合物C1强于C2,由此推测配合物C1中,U与配体中的N2O2原子配位形成共轭环,增加了分子的共轭度,降低了分子振动,增加了配合物C1的刚性,导致荧光增强。

图12 配合物C1和C2的荧光谱图Fig.12 Fluorescence spectra of complexes C1 and C2
2.7 量子化学研究
配合物C1和C2的前沿轨道HOMO和LUMO见图13和14。根据分子轨道理论,前沿轨道和邻近分子轨道对于配合物的稳定性起着重要作用[24]。对配合物分子轨道进行分析可知,HOMO与LUMO之间的能量差距越大,配合物的动力学稳定性越强。对于配合物C1和C2,其HOMO的能量分别为-0.211 5和-0.234 31 a.u.,而LUMO的能量分别为-0.100 6和-0.097 28 a.u.,2个轨道之间的能量差ΔE分别为0.110 7和0.137 03 a.u.(3.02和3.73 eV)。这一结果表明配合物C2的动力学稳定性强于C1,这是由于两者的配位构型与配位环境不同。
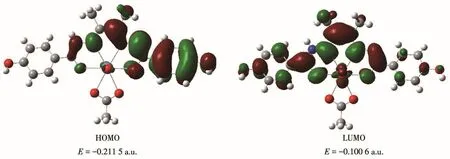
图13 配合物C1的前沿分子轨道示意图Fig.13 Schematic diagram of frontier molecular orbital for complex C1
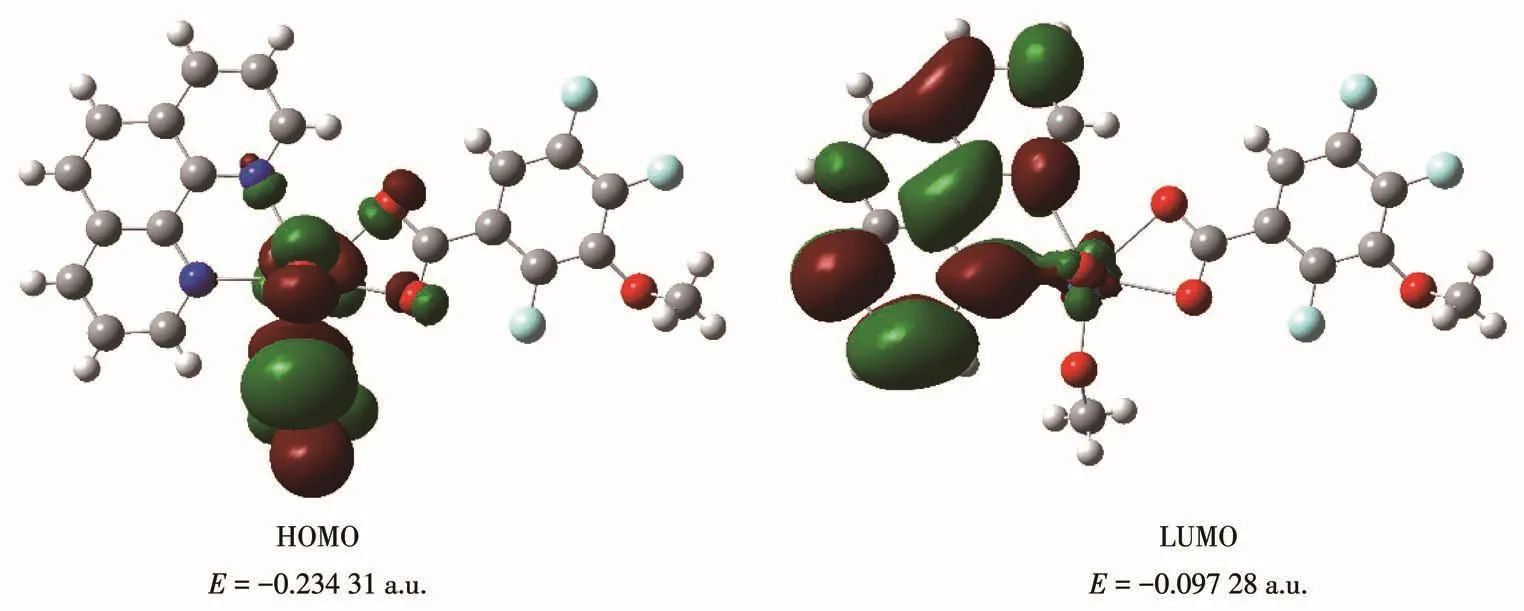
图14 配合物C2的前沿分子轨道示意图Fig.14 Schematic diagram of frontier molecular orbital for complex C2
3 结论
在溶剂热条件下,通过有机配体自组装合成了2个新的铀酰配合物C1、C2,结构分析表明,2个配合物中的U均为+6价,C1通过丰富的O—H…O氢键作用形成一维无限链状结构,C2中存在π-π堆积作用。热稳定性测试表明,配合物C1中含有溶剂分子,但是其骨架结构比配合物C2稳定。UV-Vis光谱表明配合物C1、C2均有较强的吸收带;荧光光谱表明配合物C1、C2分别在410和310 nm波长的激发下,在466和382 nm处产生明显的发射峰。另外对其结构进行量子化学计算的结果表明,配合物C2的动力学稳定性比C1强。
