Olmsted综合征一例基因突变分析
2021-12-08刘晓雁
邓 维,苏 伟,刘晓雁
掌跖角化病是一组遗传异质性的皮肤病,而Olmsted综合征(Olmsted syndrome,OS)是其中一种罕见的类型。OS的临床特征表现为残毁型掌跖角化和腔口周围角化性皮损。1927年Olmsted首次报道在1例意大利裔美国年轻男性患有严重的掌跖角化、假指骨、弯曲增厚的指甲和口唇角化过度及口角裂隙。至今超过50例OS临床患者已被报道[1-10]。近年来国内外对于OS致病基因的探索取得了重大进展。2012年林志淼等[1]率先报道了在6个OS综合征家族中发现了3个TRPV3错义突变位点:p.Gly573Ser、p.Gly573Cys和p.Trp692Gly。所有患者在1岁之前均有不同程度的掌跖角化过度,角膜过度角化,脱发和严重的病变疼痛和瘙痒。TRPV3基因又称瞬时受体离子通道3基因(transient receptor potential vanilloid 3,TRPV3), 其 在 角 质 形 成 细胞中高度表达,可编码电压和热敏离子通道。将TRPV3突变体的cDNA转染到HEK293细胞,证实这些突变都是功能获得性突变[1]。在此之后,间断有TRPV3基因的新突变被报道并证实与OS发病相关,包括 p.Gly573Ser、p.Gly573Ala、p.Trp692Cys、p.Leu673Phe、p.Gly568Asp、p.Gly568Cys, p.Gly262del、p.Gln580Pro 和 PGly568Val[2-10]。
笔者收集了1例临床表现考虑为OS的5个月龄患儿,为明确诊断及遗传咨询,行患儿全外显子基因测序,并对其父母外周血DNA行一代测序验证,发现了TRPV3基因c.1703G>T p.Gly568Val错义突变,并排除父母携带该突变,确定为散发病例。目前p.Gly568Val致OS发病国内尚无报道,现将相关结果报告如下。
1 材料与方法
1.1 临床资料
患儿,男,5个月龄。因双手指尖、足跖、臀部、外耳道及口周角化性黄红色斑块伴皲裂、脱屑4个月,于2018年10月18日就诊。患儿出生后1个月,双手指尖、双足跖部出现数个粟粒至蚕豆大小黄色角化性丘疹,部分融合成硬币大小斑块,双侧外耳道弥漫红斑,伴脱屑,唇部干裂,口角裂隙,双手指尖和双足跖斑块逐渐增厚并脱落,但仍陆续有新发皮损。患儿系第1胎第1产,出生后奶粉喂养,无特殊食物、药物过敏史,父母非近亲结婚,家族否认类似疾病史,否认遗传病史。体格检查:生长发育正常,视力、听力、智力检测无异常,系统检查无明显异常。口周可见黄色角化性斑块,双侧外耳道可见黄色黏稠性痂。双手指尖红肿,可见角化过度伴片状脱屑,肛周、双侧足跖可见黄色角化性斑块。十指(趾)甲板变薄、弯曲、部分断裂(图1)。头发、眉毛稀疏,易折断。口腔黏膜未见异常。实验室及辅助检查:外周血白细胞计数 12.88×109/L[正常值(4.00~ 10.00)×109/L],中性粒细胞绝对值 6×109/L[(2~7)×109/L)];肝肾功能、尿常规正常;双手足关节X线检查未见明显异常。根据患者临床表现,初步诊断:Olmsted综合征待排。
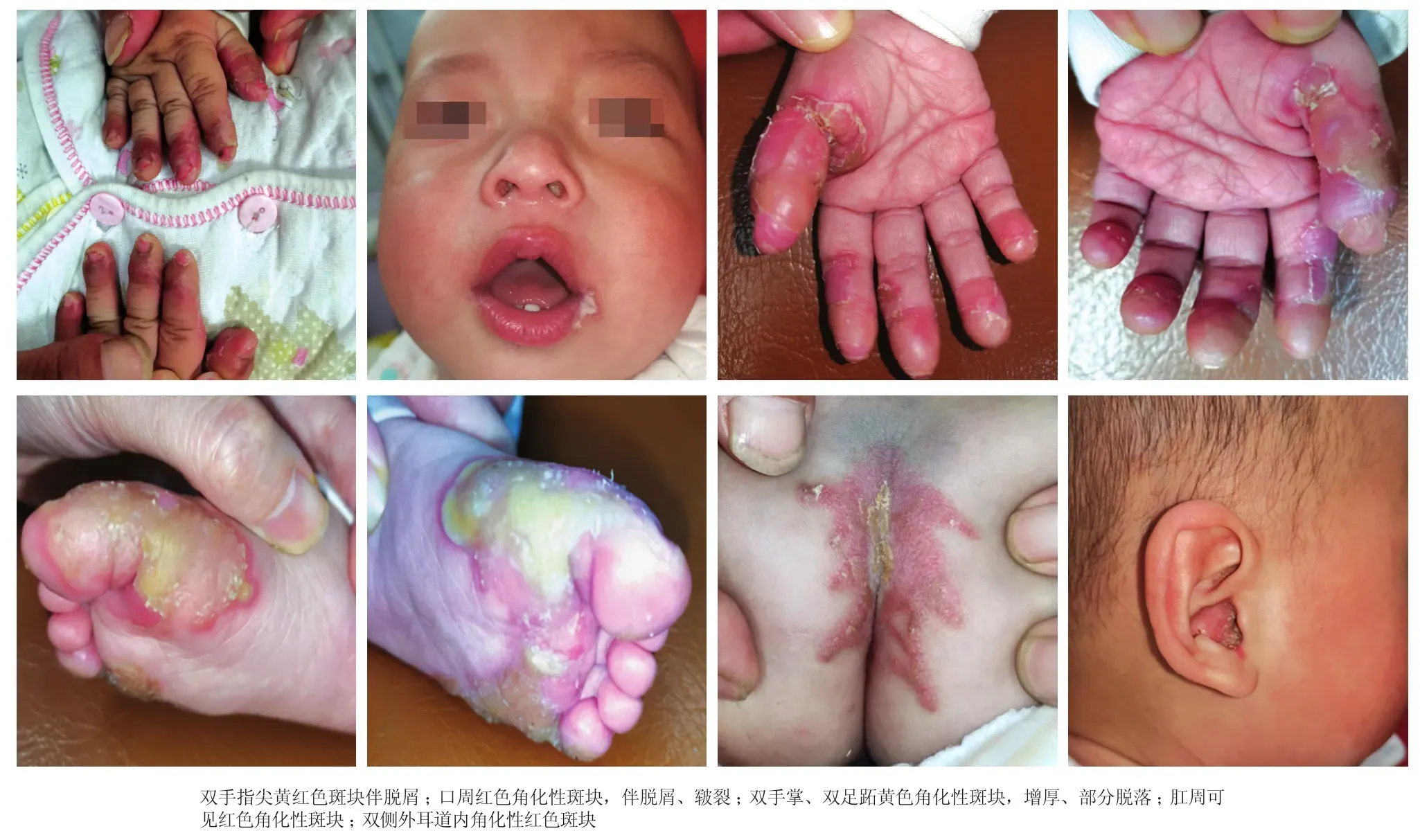
图1 Olmosted综合征患儿临床表现
1.2 目标基因捕获及高通量测序
经医院伦理委员会同意,患者及家属签署知情同意书,抽取患儿及其父母外周血各约2 ml(EDTA抗凝),用QIAamp全血DNA提取试剂盒(德国Qiagen公司)按说明书提取基因组DNA。委托北京迈基诺公司应用GenCap液相捕获目标基因技术,捕获目前已经定位全部基因的编码外显子区域。利用Illumina Nextseq 500第二代测序仪测序。用GATK软件对单核苷酸多态性(single nucleotidepolymorphisms,SNPs)和插入缺失突变(InDels)等数据进行统计和分析,筛选得到SNPs及InDels频率<0.05,且经多个数据库预测结果均为致病性的位点作为与疾病相关的候选位点。
1.3 Sanger测序验证
根据需要测序的DNA片段合成引物,用聚合酶链反应(polymerase chain reaction,PCR)法进行扩增,用ABI3730xl测序仪(美国Applied Biosystems公司)以Sanger测序法进行测序,测序结果使用Mutation Surveyor软件与参考序列进行比对分析。患者目标区域平均测序深度为153.55,99.92%目标区域覆盖度,其中>4×目标区域覆盖度为99.68%,>10×目标区域覆盖度为99.27%,>20×目标区域覆盖度为98.31%。
2 结果
TRPV3基因突变分析:13号外显子c.1703G>T杂合突变(编码区第1703号核苷酸由鸟嘌呤变异为胸腺嘧啶),导致氨基酸改变 p.G568V(第568号氨基酸由甘氨酸变异为缬氨酸),为错义突变。c.1703G>T位点未在千人基因组、ESP6500si、ExAC_ALL、ExAC_EAS及迈基诺内部1 000名正常人群数据库中发现,SIFT、PolyPhen_2、REVEL分别预测为有害(disease causing)、有害、有害,GERP++预测位点位于保守区域,且该功能域报道该区域编码瞬时受体离子通道II;该结构域存在于瞬时受体离子通道(Trp)蛋白家族中。该位点已有HGMD商业版数据库报道与OS相关[9],经家系验证患儿父母该位点无变异,患儿属于自发突变(图2)。TRPV3基因为常染色体显性遗传方式,经测序分析发现患儿TRPV3基因不遗传自父亲与母亲,符合其家族史。最后诊断:Olmsted 综合征。

图2 Olmsted综合征患儿及其父母TRPV3基因突变Sanger测序图
3 讨论
OS的报道目前多数为散发病例,好发于男性(男女比例5:1)[3,4]。OS主要表现为手、足、口腔周围严重角质化,头发稀疏以及严重的病变疼痛和瘙痒。其发病机制尚不清楚,目前人们认为突变后的TRPV3通道持续开放,导致胞内Ca2+浓度升高,进而诱导皮肤角质形成细胞凋亡从而促进了OS的发生[1]。TRPV3基因在皮肤角化、毛发生长以及瘙痒机制中发挥重要的作用。OS遗传方式近年来已被明确为常染色体显性遗传,但常染色体显性遗传患者的异常性状表达程度可有不同,且致病基因也可有一定自然突变率。本病诊断主要依靠特征性的临床表现和基因检测,组织病理改变缺乏特异性。临床上本病需要与发生于婴儿期的遗传性角化过度性疾病相鉴别,主要包括进行性遗传性手足角化病(Meleda病)和Vohwinkel残毁性遗传性角皮病(掌跖VlI型外胚叶发育不良)。三种疾病均有肢端伸侧受累、红斑边界清楚的特点,但Meleda病和Vohwinkel综合征常伴有多汗症,无腔口周围角化过度;而OS有腔口周围角化过度,但不伴出汗增多。分子遗传学上,Vohwinkel综合征为常染色体显性遗传,致病基因位于Iq21,由编码兜甲蛋白的loricrin基因突变所致。Meleda病属于常染色体隐性遗传,致病基因为SLURP-1。Vohwinkel综合征还容易合并全身性疾病,如肌病、感觉神经性耳聋等。这些疾病临床表现有时相似,则需进行基因检测协助明确诊断。
2015年,TRPV3基因c.1703G>T, p.Gly568Val错义突变被首次报道在1例7岁巴西OS女性患儿的基因检测重发现,但并未证实其是否为获得功能的突变[9]。随后2017年Nagai等报道新发现1例日本OS患者也出现Gly568Val错义突变,且其通过计算机模拟数据分析实验确定其为致病性突变[10]。本例患儿为目前发现的该基因突变位点所致的第3例OS患儿,也进一步证实了c.1703G>T, p.Gly568Val错义突变与OS致病的相关性。通过检索目前已有的20余篇TRPV3基因突变与OS的相关报道发现,不同的OS患者即使TRPV3基因突变位点相同,其发病年龄、皮损分布范围、皮损严重程度等临床表型也不尽相同,可能环境因素和(或)修饰基因在其中也发挥作用:目前已报道的具有p.Gly568Val错义突变的2例患儿相比,前者为18月龄发病的巴西患儿,起病较早,仅有局灶性和轻中度掌跖角化,无嘴唇干燥、口角皲裂及耳受累;后者为11岁发病的日本患儿,起病较晚,却患有弥漫性和严重的掌跖角化病,未提及口周、耳道受累。而本报道的患儿在出生后1个月即发病,为目前报道的最早发病的患儿;其全身皮损广泛且严重,并伴有明显瘙痒症状。造成这些个体差异的原因目前仍不明确,因此TRPV3基因型与表现型之间的关系需要进一步研究。
本例患儿的治疗目前应用维A酸乳膏剥脱角质、糠酸莫米松乳膏抗炎,夫西地酸乳膏预防感染,润肤剂保湿等外用对症治疗,但皮损无明显缓解,瘙痒症状无改善,未来皮损进展或缓解趋势无法预期。近年来随着对OS发病机制的了解加深,选择性靶向TRPV3基因治疗为角化性皮肤病的治疗提供了全新的思路。Glenmark制药公司在临床炎症与神经疼痛模型中研发出一种TRPV3拮抗剂GRC15300,GRC15300已通过1期临床试验,顺利进入后续试验研究阶段。随着TRPV3通道特异性激动剂与拮抗剂的相继研发,可能为TRPV3通道相关的遗传性角化过度性皮肤病的治疗提供一种很好的解决方案。
