钩吻素子的合成研究进展
2021-12-03刘雪艳
刘雪艳
福建医科大学药学院药物化学系,福建福州 350122
钩吻素子(koumine),见图1,是赵承嘏先生1931 年从草药钩吻(gelsemium elegans benth)中分离出来的,并确定分子式为C20H22N2O[1]。 到1981 年,利用核磁共振、X-射线单晶衍射分析以及化学降解法等手段确定了其精准的立体结构[2-3]。 可以发现,钩吻素子是一个由6 个环骈合而成的“笼状”假吲哚型生物碱,具有6 个手性碳原子(编号为C3、C5、C15、C16),其中包含两个全碳季碳中心(编号为C7、C20)。 这种特异性的结构轮廓赋予了钩吻素子独特的生物活性。

图1 钩吻素子与其晶体结构
作为胡蔓藤全草钩吻提取物的主要单体成分,钩吻素子表现出众多的药理活性。 例如可以治疗癌性疼痛和长期疼痛,且治疗指数大,无吗啡的成瘾性与耐受性;可以通过调控细胞周期实现抗肿瘤的目的;可以增强巨噬细胞吞噬能力,促进机体免疫调节;可以降低类风湿性关节炎的抗体产生,改善肿胀、痛敏、关节炎指数等症状;另外对银屑病,抗血小板聚集的治疗有一定作用[4-7]。 新近研究表明,钩吻素子可显著改善肝细胞的脂肪样病变,可用于非酒精性脂肪性肝炎的治疗等[8-11]。另一方面, 钩吻素子也是钩吻总生物碱中的毒性较低的一种,如小鼠皮下注射钩吻素子LD50 为99.0 mg/kg,仅为总碱的1/58(总碱LD50 为1.70 mg/kg)。 广泛的生理活性与相对较弱的毒性, 使得钩吻素子具有创制新型药物的重大潜能。
目前,钩吻素子主要通过从植物中提取得到,如先利用溶剂浸渍法提取钩吻总碱,再通过柱层析(或高速逆流色谱)分离制备[12-13],这种方法耗时长、步骤繁琐,收率极低,严重制约了钩吻素子的后续研究与临床应用。
利用化学合成或仿生合成一直是解决天然药物来源不足的重要手段[14-15]。 例如抗疟药物青蒿素、抗肿瘤药物紫杉醇以及抗老年痴呆症药物石杉碱甲等, 这些天然药物的人工合成是它们上市推广并造福人类的重要前提。 基于此,该文对钩吻素子的半合成、全合成以及关键中间体合成进展进行综述, 并详细阐述了其手性中心的构建, 以期促进钩吻素子及其类似物相关研究的进步。
1 钩吻素子的半合成
在钩吻素子结构被阐明清楚的基础上,Lounasmaa等[16]人提出了钩吻素子生源合成假说。他们认为钩吻素子可能是通过Hydroxygardnutine 1(羟蓬莱葛碱,一种马钱科植物蓬莱葛的提取物)转化而来(图2),可以通过分子内关环,一步构建C7、C20 2 个全碳季碳中心得到目标产物。 之后,基于这一生源合成假说,两组分别完成了钩吻素子的半合成。

图2 Lounasmaa 等提出的钩吻素子生源合成假说
刘铸晋等[17]利用老刺木碱2(Vobasine,结构上与钩吻素戊同属于蛇根精类生物碱) 为起始原料, 通过LiAlH4 还原得到老刺木二醇3 (Vobasindiol), 之后在40%稀硫酸与SeO2/H2O2体系中,一步完成脱水、烯丙基氧化(riley oxidation)和骨架重排,通过两步反应,获得了钩吻素子,从而第一次实现了钩吻素子的半合成(图3A)。 这一工作不仅验证了上述生源合成假说,同时揭示了钩吻素子与老刺木碱或钩吻素戊之间可能存在着的生源关系, 也为后来钩吻素子的合成提供了积极的借鉴意义。 尽管如此,该反应仍存在不足之处。 从反应机理上看(图式3B),该路线对可能涉及的立体选择性几乎没有加以控制, 例如利用LiAlH4 还原C3 位羰基时会产生一组对映异构体;C7、C20 成键形成2 个全碳季碳中心时可能会产生二组非对映异构体等。 这些异构体的产生可能也是目标分子总收率不高(小于25%)的原因。
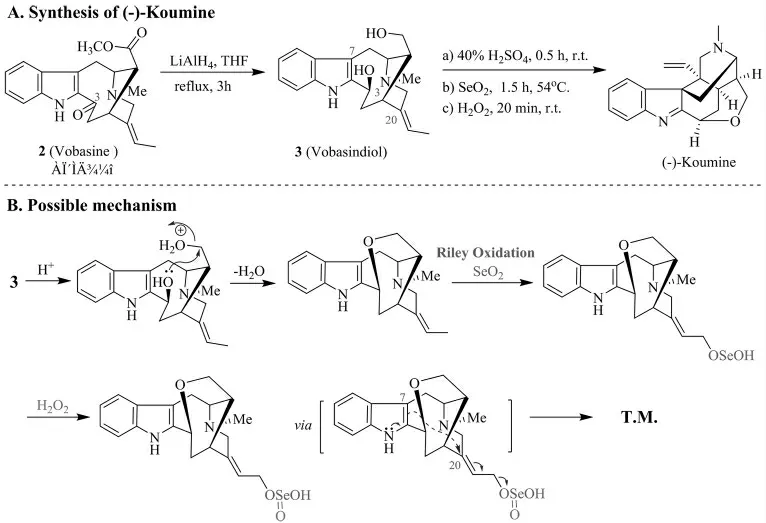
图3 刘铸晋等对(-)-Koumine 的半合成(A)及其可能的反应机理(B)
Takayama 等[18]完全按照Lounasmaa 等人提出的生源合成假说, 以天然产物Hydroxytaberpsychine 1 为起始原料,在经LiAlH4 还原后,五元氧环在3 位开环得到羟基类衍生物, 然后在吲哚母环上N 原子引入苄基保护基以及对侧链羟基进行酯化保护得到4。 4 在AlCl3 的作用下,经过芳甲醚去甲基化反应得到5,后在三氟磺酸酐的作用下,羟基转化为三氟磺酸基,在钯催化剂的体系下,脱除三氟磺酸基得到8。至此,经过6 步反应成功去除吲哚母环上的甲氧基。 对吲哚母环脱除苄基保护基后,在强碱性条件下,通过醋酸钯和三苯基膦体系中一步催化完成C7 和C20 的高效环合,得到了目标分子,总收率为21%(图4)。
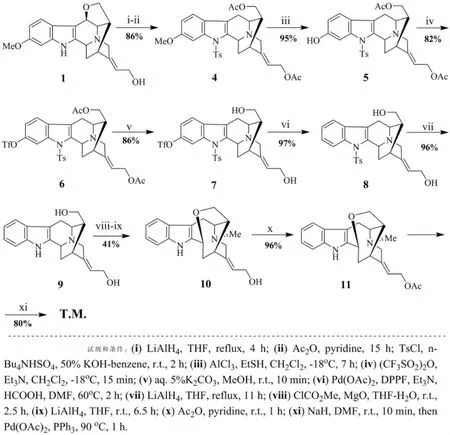
图4 Takeyama 等对(-)-Koumine 的仿生合成
令人印象深刻的是, 在该合成中前驱体11 经C7与C20 发生分子内环合, 以优异的立体选择性和较高的收率获得了钩吻素子。 首先,对这一过程运用量子化学半经验分子轨道(MNDO)理论计算方法,发现11 具有两种构象: 舒展构象的稳定态11 s 与折叠构象的不稳态11 u。两种构象的能量差仅为5.5 kcal/mol,因此在普通加热的条件下就能实现两种构象之间的快速地相互转化(图5)。 另外,C7 与C20 之间的距离间隔小于2.5A。,这为环合提供了便利。

图5 钩吻素子前驱体的构象及立体控制合成
最后,在烯丙位引入易离去基团,如碳酸酯基、酯基、磷酸酯等,在强碱性三苯基膦体系中,过渡金属醋酸钯氧化加成离去基团离去,产生一个稳定的π-烯丙基金属中间体,具有优异的立体选择性。 该方法极大的提高了整体的合成率, 且环合方法也成为后续众多钩吻素子全合成的标准方法。
生源合成的步骤具有简单高效的优点, 但是生源合成的原料均是来源于天然产物, 仍然不易获得,因此,按照刘铸晋和Takayama 等生源合成路线均难以对钩吻素子进行克级规模的制备。 而通过生源合成过程来指导化学合成天然产物的策略是一种高效的途径,同时按照可能的生源途径设计完成天然产物的合成,也为生源合成提供证据。 面对绿色化学和日益增长的需求等问题, 利用简单易得商品化的原料对钩吻素子的全合成具有更为积极的意义。
2 钩吻素子的全合成研究
Magnus 等[19]以色氨酸[(S)-(-)-Tryptophan]为起始原料完成了(+)-Koumine 的全合成(图6)。 Magnus 等首先对吲哚环N 原子上进行苄基保护基后对羧基侧链酯化得到14;14 与苯甲醛进行缩合制得15,在NaBH4 进行还原胺化制备16。 16 利用Cooks 等人发展的Pictet-Spengler 环合方法, 在苯的介质中与2-氧代戊二酸回流, 制得了一对以吲哚环为母核的三环羧基消旋体化合物17;为了对消旋体进行拆分,对羧基进行酯化后,通过柱层析即可分离得到一对非对映异构体18,19(dr.2:1)。 18 经Dieckmann 环合,构建了四环化合物20,该步反应的速率和C-3 的构象异构化有关。 合成的下一个重要阶段是通过15 号位上N 原子烷基化后桥接一个线性C4 单位后形成喹诺啶环体系。20 在酸性条件下经过7 d 时间水解脱羧, 然后利用10%Pd/C 作为相转移催化剂,移除15 号位上的苄基保护基制得21,随后与溴丙炔在乙醇的介质中进行N-烷基化反应得到丙基胺22。 22 在碳基处引入叔丁基三甲基硅酯保护基得到23;在强碱性条件下,在丙炔上引入甲氧甲酰基,成功构建了线性C4 单位,随后在去除保护基的状态下应用LiBF4 构建酮酯25。 25 在TFA/Pyrrolidine 的体系中促进分子内Michael 环化制备一组非对映异构体26/27;Tebbe 试剂将26/27 碳基还原为烯烃得制得28/29。 28/29 进行还原胺化是极为困难的, 所以应用异戊二烯在四氢呋喃介质中构建手性季碳中心。 最后经在强碱性条件下,分子内环合制得(+)-Koumine(图6)。

图6 Magnus 等对(+)-Koumine 全合成
该路线通过色氨酸侧链的手性季碳原子和Pictet-Spengler 环合构建了两个手性季碳中心,完成了对(+)-Koumine 的全合成;在最终C7, C20 环合时,发现互为非对映异构体的36/37 均能在相同条件下生成 (+)-Koumine,因此在建立喹诺啶环体系产生的顺反异构体均能环合生成目标产物, 从而避免了分离顺反异构体降低总产率。
针对Magnus 等合成路线过于冗长的问题,Bailey等[20]人进行了对钩吻素子的前驱体合成步骤进行优化,以化合物39 为起始原料,分别对11 号位和吲哚母环N原子进行保护后, 经过Dieckmann 环合反应制得了41,41 在去除苯乙酸甲酯保护基后制得了化合物42(图7)。 相比于Magnus 等的合成方法,化合物42 是合成(-)-Koumine 的前驱体, 且极大地简化了前驱体蛇根精骨架的合成步骤。
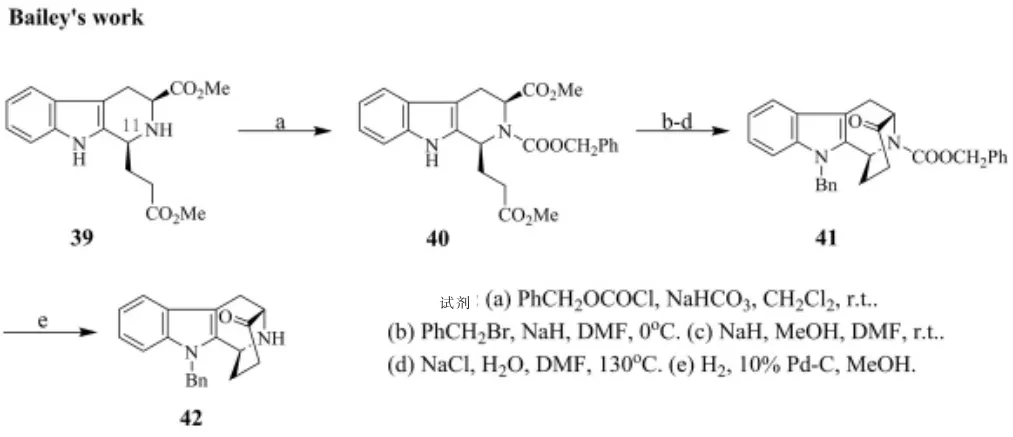
图7 Bailey 等对(-)-Koumine 全合成前驱体合成
Takayama 等[21]以简单易得的商品化原料氮杂二环壬烷43, 经过17 步反应, 以总产率7%得到 (-)-Koumine (图8)。 首先,43 在BH3.THF 的介质中进行Swern 氧化制得44;随后对羟基进行酯化保护后去除N原子上苄基保护基制备46;46 经N 原子上偶联C4 单位后与三异丙基硅基三氟甲磺酸酯反应得到48。 48 通过一价金络合物做催化剂,利用6-exo-dig 环合机理一次性构建两个手性季碳中心,制得所需产物49;同时也发生7-endo-dig 环合机理生成另一个副产物50。 值得一提的是,运用三苯甲基醚作为羟基保护基(OTr)相比于甲氧基甲基醚(MOM)具有优异的反应选择性,提高目标产物的产率(MOM 保护基: 50% yield; Tr 保护基:85% yield)。 49 在羟基脱除保护基的情况下, 利用Tebbe 试剂进行Swern 氧化获得52, 对52 母环上的羟基进行还原后去除三苯甲醚保护基制备54。 54 利用Ficher 吲哚合成法制备化合物55, 然后使用与烯烃反应具有高区域选择性的9-BBN 进行氢化还原后,在C17 处引入正确构象的羟基得到56。 最后利用了Sakai组开发的C7 和C20 的高效环合方法,得到了目标产物(-)-Koumine。

图8 Takayama 等对(-)-Koumine 全合成
该路线运用了逆合成分析的方法,一方面,利用一价金配合物进行6-exo-dig 环合,一次性建立了两个全碳季碳手性中心,高效构建(-)-Koumine 哌啶环季碳手性原子的正确构象;另一方面,利用9-BBN 的反应区域选择性构建了正确的羟基。 同时该工作还可在吲哚环上构建取代基,对(-)-Koumine 的结构改造就有很好的借鉴意义。
Kerr 等[22]发展了分子内硝酮[3+2]以及路易斯酸介导的环合反应来制备(-)-Koumine(图9)。 Kerr 组从起始原料60 开始,首先在钯催化体系下进行Still 耦合反应生成化合物61,再利用铜催化乙烯基溴化镁与61 共轭加成,生成内酯62,但是该反应并未完成乙烯基的不对成组装。 值得注意的是,使用非手性配体72 能提高第二步反应的收率和再现性, 特别是在放大反应的情况下。 利用LiAlH4 还原内酯62,制得二醇63;对63 烯丙醇基团进行区域选择性取代并对羟胺进行保护得到单一异构体64。 64 与吲哚衍生物73 进行组装后,生成合成中间体65,随后在苯的介质中进行回流,进行分子内硝酮[3+2]环化,获得一组非对映异构体66,67。 为了进行拆分工作, 在混合状态下用镁去除对甲苯磺酰基保护基团,生成无保护的吲哚母核;随后对侧链上的羟基进行Swern 氧化生成醛类化合物,最后需对醛基进行保护,以避免SmI2 还原。 自此,经过3 个步骤后顺利拆分制得69。 69 在路易斯酸BF3.OEt2 与乙腈的体系中,经过Pictet-Spengler 环合反应,羟基与吲哚母环8 号位上经分子内关环构筑手性季碳中心得到化合物70。 最后利用了刘铸晋的方法, 实现了 (-)-Koumine 的全合成。

图9 Kerr 等对(-)-Koumine 全合成
学者对双环辛烷骨架的合成产生极大的兴趣,围绕酮α-烯丙基化构建双环辛烷骨架这一目标展开了大量的研究工作,如探索SN2 反应条件等,其研究成果对后续钩吻素子全合成的发展也具有一定的借鉴意义[23-27]。
3 中间体骨架的合成
徐风组等[28]对具有19-C,20-C 双键,16-C 氧代的蛇根精骨架进行合成, 他们的策略是通过立体专一性合成1,2,3,4-四氢-β-咔啉, 然后再通过自由基环化反应,完成具有正确取代的蛇根精骨架的合成(图10)。从L-色氨酸甲酯74 出发, 与3-甲氧碳基丙酰氯在吡啶中酰胺化得到75。 75 在三氯氧磷中进行Bischler Napieralski 反应,成功得到了3,4-二氢-β-咔啉76。 由于76 在NaBH4 还原体系中极不稳定, 因此采用PtO2/EtOH 体系中常压催化氢化立体专一性得到1,2,3,4-四氢-β-咔啉77,产率为99%。 得到77 后,用1-溴-4-乙酰氧基-Z-丁烯-2 与77 反应,顺利的得到了78。 经过13C 波谱分析法对77 的检测也确证催化氢化反应的立体专一性。 再用乙烯基乙醚保护78 吲哚氨基,得到79。79 经Dieckmann 反应得到β-酮酯80。在室温条件下经Mn(Ⅲ)/Cu(Ⅱ)自由基环化反应得到81。 由于81 的15 位羟基对酸对热不稳定,采用酯化的方式保护15 位的羟基, 再在HOAc/MeOH/H2O 的体系中除去吲哚保护基,得到83。 83 的合成克服了合成蛇根精骨架分子所遇到杂环高张力的问题。

图10 徐风组对于蛇根精骨架的合成
近年来, 学者开发探索钩吻素子中间体的高效合成策略,以促进钩吻素子的全合成发展(图11)[29-33]。 以钩吻素子中间体85 为分支点, 在C3-N 键断裂后,通过π-Lewis 酸催化仿生吲哚加成到丙二烯单元, 进而构建钩吻素子支架。 因而,具有通用酮和丙二烯基团的高级笼中间体——氮杂双环辛烷85 为合成钩吻素子的核心步骤。 具有指示手性碳中心的84,可以以L-色氨酸为起始原料,发生kulinkovich 环丙烷化反应生成。然后,84 通过串联胺氧化和环丙醇开环环化快速组装氮杂双环壬烷骨架并引入通用酮基团。 氮杂双环壬烷部分可通过迪克曼环化、烯烃复分解、吲哚基Friedel-Crafts 反应、和[5+2]-环加成或环扩大合成,氮杂双环辛烷部分最常通过钯催化偶联过程通过酮的α-乙烯基化构建,为了增加后期结构多样化,在C20 处引入更通用的丙二烯团体[34-38]。
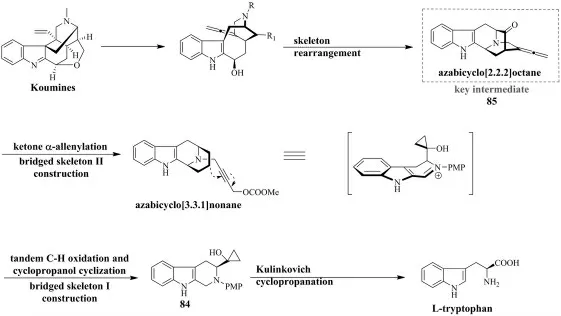
图11 钩吻素子中间体-氮杂双环辛烷的逆合成
综上所述, 该文对钩吻素子的合成进展进行了全面综述,包括半合成路线、全合成路线以及中间体骨架的合成路线,并详细阐述了其手性中心的构建,以促进钩吻素子以及相关衍生物的合成和研究。
