(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸衍生的手性咪唑啉酮催化剂合成新方法
2021-12-02黄志诚李臣毅李文升王立新
王 毅, 黄志诚, 向 敏, 李臣毅, 张 健, 李文升, 田 芳, 王立新
(1. 中国科学院 成都有机化学研究所,四川 成都 610041; 2. 中国科学院大学,北京 100049)
咪唑啉酮催化剂是由MacMillan小组开发的一种手性仲胺类咪唑啉酮催化剂,常与酸形成相应的盐来催化反应。这类催化剂能够通过烯胺与亚胺离子机理催化α,β-不饱和醛酮类底物的1,3-偶极环加成[1]、Diels-Alder反应[2]、不对称Michael加成[3-4]以及吡咯与吲哚类底物的不对称傅克烷基化[5]反应;也能通过SOMO机理对反应底物进行活化[6],催化醛酮类底物发生α-位卤代反应[7-8],转移氢化反应[9-10],以及醛类底物的α-位烷基化[11]等;还可以通过协同催化机理与金属离子共同催化醛类底物的α-位芳基化[12],催化烯烃与醛类底物制备吡咯烷类化合物[13]等。综上所述,咪唑啉酮类催化剂在不对称催化领域之中被广泛应用,是一类非常重要的有机小分子催化剂。

Chart 1
咪唑啉酮类催化剂可以由各类氨基酸衍生得到,常见的几种咪唑啉酮催化剂如下:
这类催化剂合成的步骤相对简单,一般先将相应氨基酸酯化,随后在甲胺的甲醇溶液中反应,得到氨基酸衍生的酰胺产物,最后再与醛酮发生缩合,即得对应咪唑啉酮催化剂[2]。2015年,Gryko课题组[14]尝试使用了上述方法以(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸为原料合成咪唑啉酮类催化剂,并将其应用于苯丙醛的α-位不对称光氧化反应之中,最高得到74%的ee值。该课题组以NaCN作为催化剂,氨基酸酯和甲胺反应耗时9 d以96%的收率得到氨基酰胺。3步反应总耗时近10 d,且合成中使用剧毒的NaCN作为催化剂,合成条件苛刻,后处理复杂,反应总收率仅为35%(Scheme1)。

Scheme 1
为改进此催化剂的合成路线,降低合成难度和成本,提出一种合成此催化剂的新方法(Scheme 2),使用相对安全的三光气代替传统的剧毒光气合成N-羧基氨基酸酐(NCA,6)[15],而后与甲胺反应,在室温下7 h即可定量得到对应(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺7,反应时间大幅度缩短,收率大幅提高,这对于简化该催化剂的合成、分离及纯化具有一定的现实意义。考察了反应溶剂、原料摩尔比和反应温度等因素对环化反应收率的影响。产物结构经1H NMR和13C NMR确证,与文献报道一致[14]。
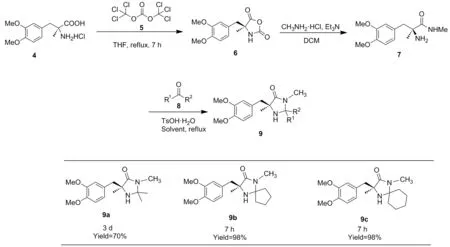
Scheme 2
1 实验部分
1.1 仪器与试剂
Bruker-300/400 MHz型核磁共振仪(TMS为内标)。
所用试剂均为分析纯。
1.2 合成
(1)N-羧基氨基酸酐(6)的合成
称取(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺0.50 g(1.8 mmol)并将其与15 mL四氢呋喃混合,搅拌使固体悬浮于体系之中。将三光气1.33 g(4.5 mmol)投入反应体系中搅拌,随后升温回流7~8 h。反应体系开始为乳白色悬浮液,随反应进行逐渐变为均相。冷却至室温,减压除去溶剂,残余物经过硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=1/1~1/2]纯化得纯白色固体60.41 g(1.53 mmol),收率85%;1H NMR(400 MHz, CDCl3)δ: 6.80(d,J=8.1 Hz, 1H), 6.70~6.63(m, 2H), 3.85(s, 3H), 3.82(s, 3H), 3.12(d,J=14.0 Hz, 1H), 2.88(d,J=14.1 Hz, 1H), 1.59(s, 3H);13C NMR(101 MHz, CDCl3)δ: 172.3, 151.6, 149.1, 148.8, 125.5, 122.3, 113.1, 111.4, 64.5, 55.9, 55.8, 43.6, 23.8。
(2) 氨基酰胺(7)的合成
称取N-羧基氨基酸酐0.50 g(1.89 mmol),将其溶解于20 mL二氯甲烷中,冰水浴下向体系中滴加三乙胺0.23 g(2.27 mmol),搅拌15 min后向其中一次性加入甲胺盐酸盐0.15 g(2.27 mmol),自然升温,搅拌约7 h后反应完全(TLC检测)。过滤除去不溶性的盐酸盐,随后依次用饱和碳酸钠水溶液(3×10 mL)洗涤,蒸馏水(3×10 mL)洗涤,收集有机相,分液除水后减压除去溶剂,得淡黄色油状液体(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺70.47 g(1.85 mmol),收率98%(纯度93%,Detected by HPLC);1H NMR(300 MHz, DMSO-d6)δ: 6.82(d,J=8.1 Hz, 1H), 6.71(d,J=1.9 Hz, 1H), 6.64(dd,J=8.1 Hz, 1.9 Hz, 1H), 3.69(d,J=4.6 Hz, 6H), 3.00(d,J=12.9 Hz, 1H), 2.61~2.36(m, 4H), 1.18(s, 3H);13C NMR(75 MHz, CDCl3)δ: 176.9, 148.5, 147.6, 129.4, 122.1, 112.8, 110.6, 58.2, 55.6, 46.3, 27.9, 25.8。
(3) 咪唑啉酮催化剂(9)的合成
9a: 称取70.47 g(1.85 mmol)溶解于10 mL甲醇之中,充分搅拌溶解后加入5 mL丙酮与TsOH·H2O 0.035 g(0.185 mmol),回流36 h后向体系中补加5 mL丙酮,随后再回流36 h,减压除去溶剂,残余物经过硅胶柱层析[洗脱剂:V(甲醇)/V(乙酸乙酯)=1/10]纯化得淡黄色油状液体9a0.38 g(1.30 mmol),收率70%;1H NMR(300 MHz, DMSO-d6)δ: 6.82(d,J=8.1 Hz, 1H), 6.76(d,J=1.9 Hz, 1H), 6.67(dd,J=8.1 Hz, 2.0 Hz, 1H), 3.68(d,J=2.5 Hz, 6H), 2.86(d,J=13.3 Hz, 1H), 2.58(s, 3H), 2.45(d,J=13.4 Hz, 1H), 1.21(s, 3H), 1.17(s, 3H), 0.75(s, 3H);13C NMR(75 MHz, DMSO-d6)δ: 174.4, 148.2, 147.5, 129.8, 122.4, 114.0, 111.2, 73.9, 62.5, 55.5, 55.4, 44.0, 28.5, 27.4, 27.0, 24.8。
9b: 称取70.47g(1.85 mmol)溶解于10 mL环己烷之中,充分搅拌溶解后加入2 mL环戊酮与TsOH·H2O 0.035 g(0.185 mmol),回流7 h后(TLC检测)。反应完毕,减压除去溶剂,残余物经过硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=1/1~1/2]纯化得淡黄色油状液体9b0.57 g(1.81 mmol),收率98%;1H NMR(300 MHz, CDCl3)δ: 6.87~6.52(m, 3H), 3.81(d,J=6.5 Hz, 6H), 3.23(d,J=13.9 Hz, 1H), 2.68(s, 3H), 2.54(d,J=13.9 Hz, 1H), 2.25~1.43(m, 8H), 1.35(s, 3H);13C NMR(75 MHz, CDCl3)δ: 175.7, 148.7, 147.8, 128.8, 122.1, 112.7, 110.7, 84.4, 63.4, 55.7, 55.6, 37.4, 36.7, 33.1, 26.2, 25.5, 23.9, 23.6。
9c: 称取70.47g(1.85 mmol)溶解于10 mL环己烷之中,充分搅拌溶解后加入2 mL环己酮与TsOH·H2O 0.035 g(0.185 mmol),回流7 h后(TLC检测)。反应完毕,减压除去溶剂,残余物经过硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=1/1~1/2]纯化得淡黄色油状液体9c,收率98%;1H NMR(300 MHz, DMSO-d6)δ: 6.81~6.51(m, 3H), 3.61(s, 6H), 2.76(d,J=13.3 Hz, 1H), 2.50(s, 3H), 2.40(d,J=13.4 Hz, 1H), 1.68~0.79(m, 13H);13C NMR(75 MHz, DMSO-d6)δ: 174.6, 148.2, 147.5, 129.9, 122.3, 114.0, 111.3, 75.3, 62.4, 55.5, 55.3, 44.1, 36.2, 35.0, 27.41, 24.9, 24.4, 22.0, 21.9。
(4)N-羧基氨基酸酐(11)的合成
称取100.50 g(3.0 mmol)并将其与15 mL四氢呋喃混合,搅拌使固体悬浮于体系之中。向体系中加入三光气2.20 g(7.5 mmol)后持续搅拌,升温回流2~3 h。反应体系开始为乳白色悬浮液,随后逐渐成为均相。冷却至室温,减压除去溶剂,冰水浴条件下向体系中加入25 mL正己烷,持续搅拌,随后有大量固体析出,抽滤得纯白色固体110.57 g,不经纯化直接投入下步反应中[16]。
(5) 咪唑啉酮催化剂(1)的合成
称取110.20 g(约1.05 mmol),并将其溶解于5 mL二氯甲烷之中,冰水浴条件下向体系中缓慢滴加30%含量的甲胺醇溶液0.16 g,体系缓慢升至室温,搅拌过夜。减压除去溶剂,不经纯化即可投入下一步反应之中。向上一步得到的产物中加入5 mL丙酮,5 mL甲醇与TsOH·H2O 0.019 g(0.10 mmol),搅拌至溶解后升温回流18 h。减压除去溶剂,残余物经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=1/2]纯化得无色油状液体10.18 g(0.84 mmol),收率80%;1H NMR(300 MHz, CDCl3)δ: 7.36~7.07(m, 5H), 3.72(dd,J=6.9 Hz, 4.5 Hz, 1H), 3.08(dd,J=14.1 Hz, 4.4 Hz, 1H), 2.93(dd,J=14.1 Hz, 6.8 Hz, 1H), 2.68(s, 3H), 1.19(s, 3H), 1.09(s, 3H);13C NMR(75 MHz, CDCl3)δ: 173.4, 137.1, 129.5, 128.6, 126.7, 75.5, 59.2, 37.2, 27.2, 25.3, 25.2。
2 结果与讨论
2.1 条件优化
本合成路线共有3步,关键步为环化反应。开环反应几乎能够定量得到氨基酰胺7产物,缩合反应条件相对固定,为得到更高的收率,对环化反应的条件进行了优化,考察了溶剂,配比,温度,反应时间等因素对反应收率的影响。
(1) 溶剂对环化反应的影响
表1为溶剂对于反应的收率的影响,反应在醚类溶剂中能得到相对较高的收率(Entries 1~4),其中环醚类溶剂如四氢呋喃和1,4-二氧六环等相对于直链醚类要好,综合而言,反应的最佳溶剂为四氢呋喃

表1 溶剂对反应Ⅰ收率的影响
(2) 原料摩尔比对环化反应收率的影响
在确定最佳溶剂为THF后,决定对原料的比例进行优化,表2为反应原料配比对反应收率的影响,随着三光气的比例逐渐增加,6的收率逐渐增加;但是继续增加三光气的比例,6的收率并未有进一步提升反而出现下降,因而可以确定反应原料的最佳比例为1/2.5。
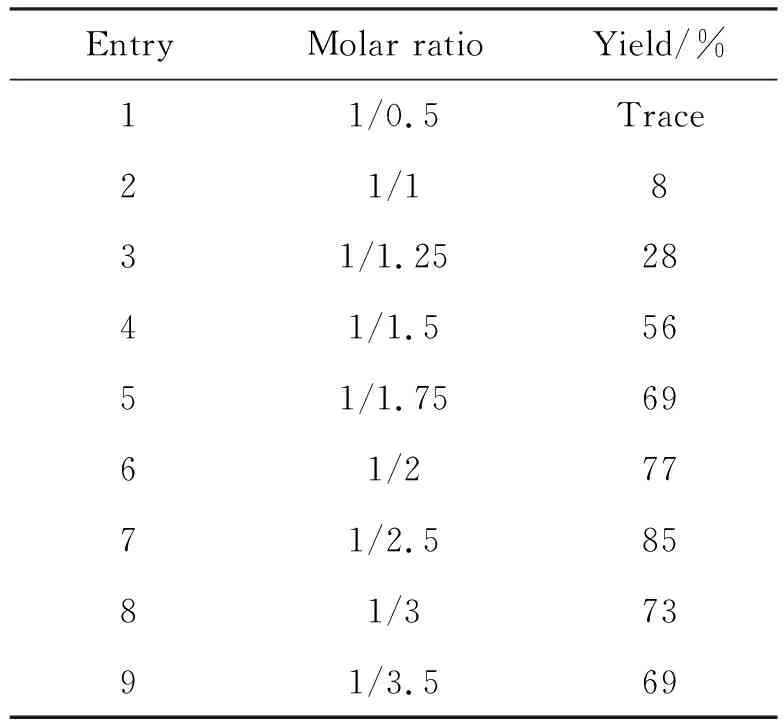
表2 原料配比对反应收率的影响
(3) 反应温度和反应时间对环化反应收率的影响
在确定最佳溶剂以及最佳反应摩尔比以后,尝试对反应温度和反应时间进行了优化,表3为反应温度和反应时间对环化反应收率的影响。反应收率随温度的上升而不断增加;从中可以看出,在回流温度下反应7 h收率就能达到85.4%,继续延长反应时间,收率的增长并不明显,因而反应最佳温度即为回流温度,反应时间为7 h(Entries 4~8)。
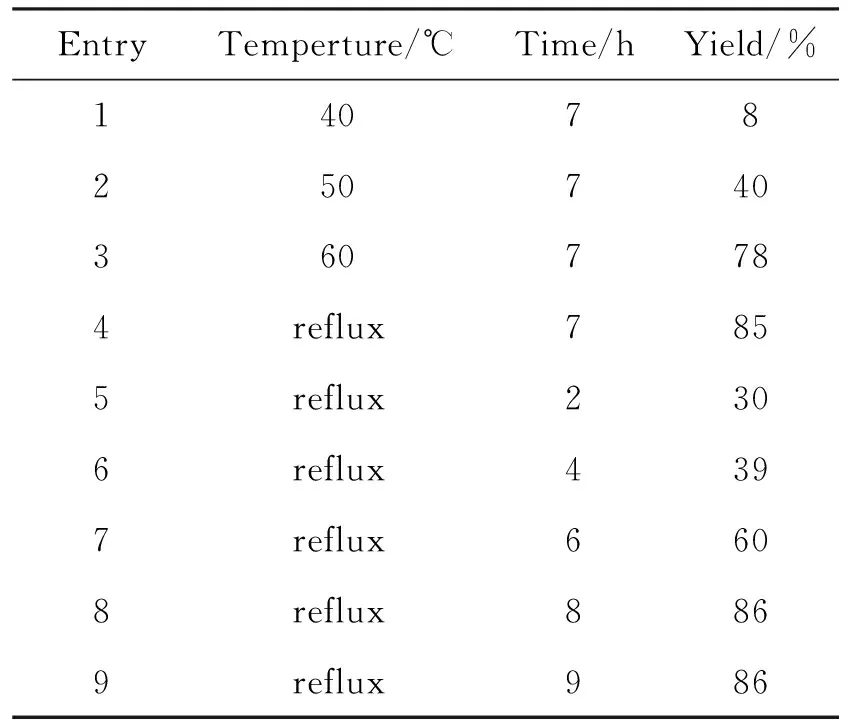
表3 反应温度和时间对反应收率的影响
综上所述,环化反应的最佳条件为(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸盐酸盐与三光气比例为1/2.5, THF做溶剂,回流反应7 h。
2.2 反应的底物拓展
(1) 咪唑啉酮类催化剂的合成
将该路线应用于苯丙氨酸制备MacMillan催化剂同样取得较好的结果,反应总收率能达到80%(Scheme3)。相对于经典合成方法仅59%的总收率[2],本路线收率有很大提高,也证明了本路线可应用于多种咪唑啉酮类催化剂的制备。

Scheme 3
(2) 缩合反应的底物拓展
对酮类底物进行了拓展,反应均使用10%当量的一水对甲苯磺酸做催化剂。当酮类底物为环戊酮时,反应耗时7 h,能够以98%收率得到环戊酮衍生咪唑啉酮9b;当酮类底物为环己酮时,反应耗时7 h,能够以98%收率得到环己酮衍生咪唑啉酮9c。
以(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸盐酸盐为原料,设计了一条合成咪唑啉酮类催化剂的新路线,并对其中的关键步环化反应进行了条件优化。相对于之前的合成路线,本路线耗时大幅缩短,过程中无需使用剧毒的NaCN催化,合成总收率最高提升至85%;且路线同样适用于合成其他咪唑啉酮催化剂。
