基于UPLC的不同产地野菊花多组分定量分析△
2021-11-16魏伟锋韩正洲魏民李建领张凯姚慧颖王信宏
魏伟锋,韩正洲,魏民,李建领,张凯,姚慧颖,王信宏,*
1.华润三九医药股份有限公司,广东 深圳 518110;2.山东农业工程学院,山东 济南 250100;3.嘉应学院,广东 梅州 514031
野菊花为菊科植物野菊Chrysanthemum indicumL.的干燥头状花序[1],为清热解毒类中成药及相关制剂的主要原料,生长于草地、田野、路边等,在民间及临床上均应用广泛[2-4]。植物化学研究表明,野菊花主要含有黄酮类、萜类、绿原酸类、叶绿素、微量元素等化合物[5-7]。其发挥清热解毒功效的活性物质主要为黄酮类和有机酸类化合物[5,8-10]。蒙花苷、木犀草素、芹菜素和木犀草苷为黄酮类化合物中的主要代表性成分,具有抗炎、保肝、抗肿瘤、保护心血管系统、抗菌等多种药理作用。绿原酸为有机酸类化合物中的代表性成分,具有广谱抗菌、抗病毒、保肝、利胆、抗肿瘤、降血压、调血脂、清除自由基等多种药理活性[3,11-12]。
中药的药效是多种化学成分共同作用的结果,仅检测蒙花苷的含量不足以准确评价和比较不同产地野菊花的质量。本研究选取了39 份不同产地野菊花样品,建立了基于超高效液相色谱法(UPLC)的野菊花定量分析方法,同时对蒙花苷、木犀草素、芹菜素、木犀草苷和绿原酸5 个化合物进行定量分析,为野菊花的质量评价提供参考。
1 材料
1.1 试药
野菊花样品均为野生样品,初花期采收,晒干后保存,产区信息见表1,经华润三九医药股份有限公司韩正洲工程师鉴定为野菊Chrysanthemum indicumL.的干燥头状花序,凭证样品存放于华润三九医药股份有限公司研发中心。

表1 野菊花样品来源信息
对照品绿原酸(纯度≥96.6%,批号:110753-201314)、木犀草苷(纯度≥94.9%,批号:111720-201609)、木犀草素(纯度≥99.6%,批号:111520-200504)、蒙花苷(纯度≥96.6%,批号:111528-201710)、芹菜素(纯度≥99.6%,批号:111901-201102)均购自中国食品药品检定研究院;色谱纯乙腈、甲醇均购自赛默飞世尔科技有限公司;其余试剂均为分析纯。
1.2 仪器
ACQUITY UPLC H-Class 型超高效液相色谱系统(美国Waters 公司);MF10B 型研磨机(德国IKA 公司);ME36S 型百万分之一分析天平(德国Sartorius 公司);XS204 型万分之一分析天平(瑞士Mettler toledo 公司);SB-400DTY 型超声波清洗机(宁波新芝生物科技股份有限公司)。
2 方法与结果
2.1 色谱条件
色谱柱:ACQUITY UPLC CSH C18(150 mm×2.1 mm,1.7 µm);流动相:乙腈(A)-0.02%磷酸溶液(B);梯度洗脱(0~10 min,10%~15%A;10~15 min,15%~25%A;15~30 min,25%~30%A;30~35 min,30%~10%A;35~37 min,10%A);进样体积:2 µL;柱温:(30±5)℃;流速:0.4 mL·min-1;检测波长:326 nm。
2.2 供试品溶液制备
精密称定过三号筛(50 目)的干燥野菊花粉末0.2 g,置于100 mL 的具塞锥形瓶中,加入70% 甲醇50 mL,称定质量,超声提取30 min(功率:500 W,频率:40 kHz),放冷,再称定质量,用70%甲醇补足减失质量,摇匀,静置,吸取提取液,0.22 µm 微孔滤膜滤过,取续滤液作为供试品溶液[13]。
2.3 混合对照品溶液制备
精密称取对照品绿原酸、木犀草苷、木犀草素、蒙花苷、芹菜素适量,用甲醇溶解并定容至100 mL,得到质量浓度分别为11.82、20.35、11.11、39.68、10.00µg·mL-1的混合对照品溶液。精确吸取混合对照品 溶液0.50、1.00、2.50、5.00、10.00 mL 于10 mL 量瓶中,用甲醇定容,即得系列质量浓度的混合对照品溶液。
2.4 系统适用性与线性关系考察
分别吸取2.3 项下的系列混合对照品溶液,按2.1 项下色谱条件进样测定,5 个成分均能达到基线分离,与相邻色谱峰的分离度均大于2.0,以各成分色谱峰计算理论塔板数>2000,且峰形较好,典型的UPLC 色谱图见图1。以对照品质量浓度为横坐标(X),以峰面积为纵坐标(Y),绘制标准曲线,见表2。结果显示,5 个成分在相应的质量浓度范围内与峰面积的线性关系均良好(r≥0.999 0)。

图1 混合对照品与野菊花样品的UPLC图

表2 各化合物线性关系考察
2.5 精密度试验
精密称取野菊花混合样品0.2 g,按2.2 项下方法处理,按2.1项下色谱条件连续进样6次,记录峰面积,计算绿原酸、木犀草苷、木犀草素、蒙花苷、芹菜素峰面积RSD 分别为0.97%、1.39%、0.65%、1.78%、1.19%,均低于2.0%。表明仪器精密度良好。
2.6 重复性试验
精密称取野菊花混合样品0.2 g,按2.2 项下方法平行制备6 份,按照2.1 项下色谱条件进行UPLC分析,记录峰面积,绿原酸、木犀草苷、木犀草素、蒙花苷、芹菜素峰面积的RSD 分别为1.67%、1.77%、1.82%、1.06%、1.98%,均小于2.0%,表明方法具有良好的重现性。
2.7 稳定性试验
精密称取野菊花混合样品0.2 g,按2.2 项下方法处理,按照2.1 项下色谱条件,分别在0、2、4、8、12、24、48 h 测定,记录峰面积,计算绿原酸、木犀草苷、木犀草素、蒙花苷、芹菜素峰面积RSD分别为1.74%、0.49%、0.13%、1.15%、1.36%,均小于2.0%,表明供试品溶液中的5 个成分在室温条件下48 h内稳定。
2.8 加样回收率试验
取已知含量的野菊花样品粉末6 份,精密称定0.1 g,按2.2 项下方法处理,分别精密加入一定量的绿原酸、木犀草苷、木犀草素、蒙花苷、芹菜素对照品溶液,按2.1 项下色谱条件下进样,记录峰面积,计算其加样回收率,结果见表3,加样回收率为99.01~99.82%,RSD最高为1.35%。
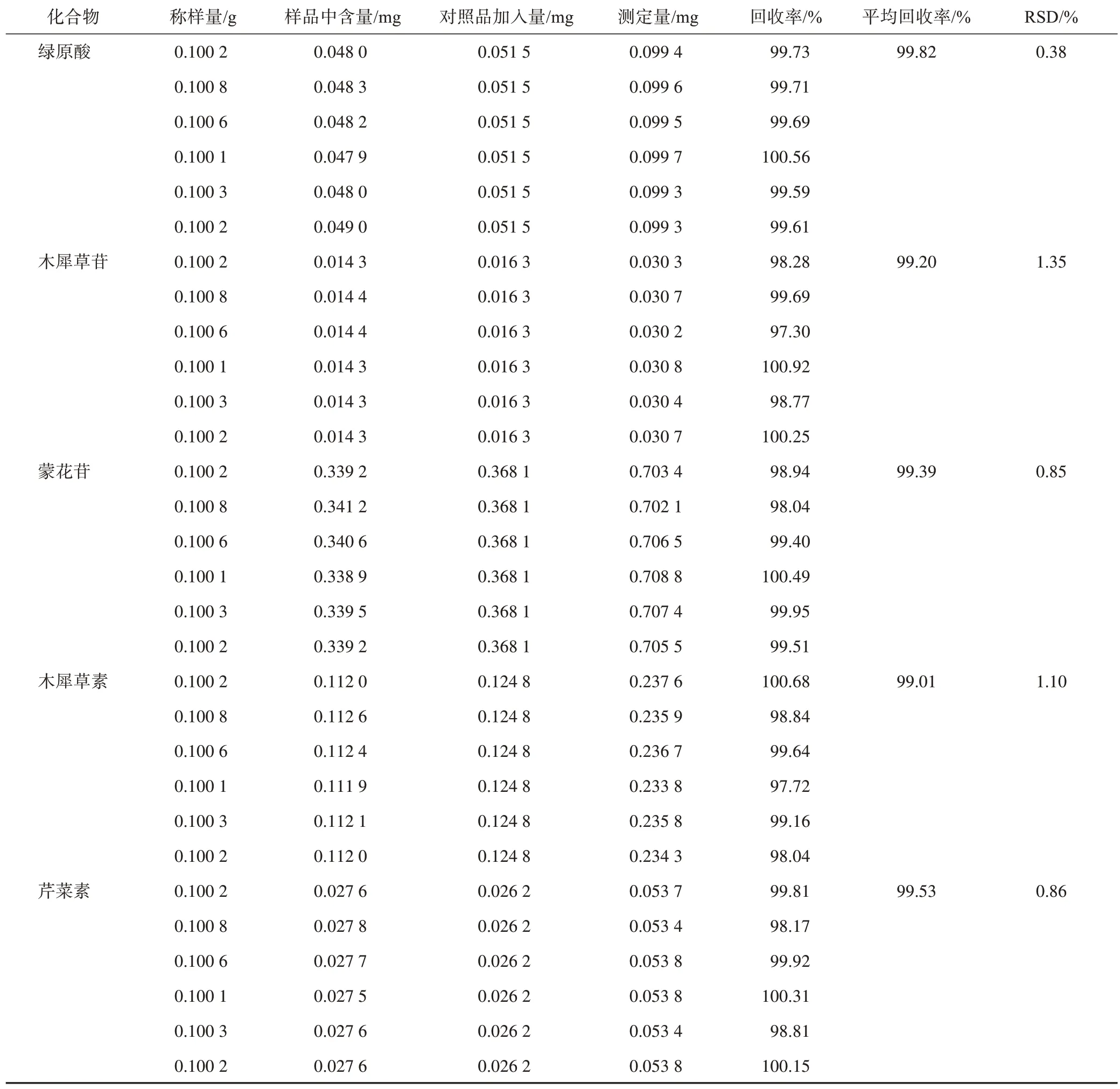
表3 野菊花5个成分加样回收率试验结果
2.9 5个成分的定量测定
将39 批野菊花样品按2.2 项下方法处理,每个批次平行3 份,在2.1 项下色谱条件下进行定量测定,计算各供试品中5 个成分的含量,结果见表4。《中华人民共和国药典》(以下简称《中国药典》)2020 年版规定野菊花中蒙花苷质量分数不得低于0.8%。实验结果表明,39 份野菊花样品中有23 份未达到合格水平,其中广东(7、8 号)、广西(11、13 号)、湖北(29、33 号)、重庆(35、36 号)、河南(15 号)地区9 份样品未检测到蒙花苷;湖北随州样品(24 号)绿原酸质量分数最高,为5.40 mg·g-1,湖北宜昌样品(27 号)绿原酸质量分数最低,为0.30 mg·g-1;湖北双牌野菊花样品(38 号)木犀草苷质量分数最高,为2.80 mg·g-1,广西南丹样品(12 号)木犀草苷质量分数最低,为0.10 mg·g-1;湖南益阳样品(39 号)木犀草素质量分数最高,为11.90 mg·g-1,湖北随州样品(22、24号)木犀草素质量分数最低,为0.60 mg·g-1;湖北监利样品(28 号)芹菜素质量分数最高,为1.10 mg·g-1,河南汝州(16 号)、河南焦作(17 号)、湖南双牌(38 号)3 份样品中未检测到芹菜素。

表4 野菊花5个成分含量测定结果 mg·g-1
3 分析
3.1 UPLC方法学研究
UPLC 具有高分离度、高灵敏度、洗脱时间短等优点[14-17]。谭晓杰等[18]建立了HPLC 测定野菊花蒙花苷的方法,检测波长选择326 nm;崔兰冲等[19]研究了超声提取野菊花组分的方法。在前人研究的基础上,本研究选择了70%的乙醇超声提取30 min 的样品处理方法,采用ACQUITY UPLC CSH C18(150 mm×2.1 mm,1.7 µm)色谱柱,乙腈-0.02%磷酸溶液为流动相梯度洗脱,流速为0.4 mL·min-1,柱温为(30±5)℃,进样体积为2µL,检测波长为326 nm。在此条件下,5 个成分线性关系良好,精密度、稳定性、重复性及加样回收率试验结果表明,此方法可以用于野菊花定量分析。
3.2 不同产地野菊花定量分析
本研究对39 批次野生野菊花进行了定量分析,绿原酸、木犀草苷、蒙花苷、木犀草素、芹菜素质量分数分别为0.30~5.40、0.10~2.80、0~15.40、0.60~11.90、0~1.10 mg·g-1。说明不同产地野生野菊花各成分含量差异显著,质量参差不齐。建立合理、有效的中药质量评价体系是实现中药现代化的前提之一,本研究的野菊花样品,按《中国药典》2020 年版规定[1],合格率仅为41.0%,同时有9 份样品未检测到蒙花苷。谭晓杰等[18]、崔兰冲等[19]的研究中也检测到大量不合格样品。多种证据表明仅以蒙花苷含量判定野菊花药材质量不够全面,仍需深入探讨。
