钙离子信号介导Nod样受体蛋白炎性小体激活的研究进展
2021-11-07徐志猛
陈 雨,徐志猛,李 萍
(中国药科大学天然药物活性组分与药效国家重点实验室,南京 210009)
Nod样受体蛋白(Nod-like receptor protein 3,NLRP3)炎性小体作为重要的固有免疫组成部分,能对包括入侵病原体、宿主细胞衍生的危险信号和环境刺激物在内的各种刺激做出反应,在机体免疫反应发生过程中起着重要作用。NLRP3炎性小体的异常激活可导致多种自身炎症或慢性炎症疾病如阿尔茨海默病、炎症性肠病和冷热相关周期性综合征的发生发展。明确NLRP3炎性小体的激活机制,对通过降低其活性而用于疾病的防治十分重要。
钙离子作为细胞中广泛表达的第二信使,对细胞内信号传导、维持细胞稳态具有重要的作用。胞内Ca2+信号控制多种细胞过程,包括增殖和分化、转录、细胞代谢和细胞死亡[1]。研究发现,在不同NLRP3炎性小体激活剂的作用下细胞都出现了胞浆Ca2+浓度升高的现象,并且抑制异常的Ca2+流动能够降低细胞白细胞介素1β(interleukin-1β,IL-1β)水平[2]。然而,对于Ca2+的变化如何激活NLRP3炎性小体仍存在着许多有待研究之处。本文就Ca2+在NLRP3炎性小体激活和调控中的作用进行综述。
1 NLRP3炎性小体的结构和激活
1.1 NLRP3炎性小体的结构
NLRP3炎性小体是细胞内的大分子复合物,包括传感器分子NLRP3、适配器凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a caspase recruitment domain,ASC)、有丝分裂激 酶NIMA相 关 激 酶7(NIMA-related kinase 7,NEK7)和效应器蛋白酶半胱天冬酶1(cysteinedependent aspartate-specifc proteases 1,caspase-1)[3]。NLRP3包含一个N端pyrin结构域(pyrin domain,PYD)、连接结构NACHT以及C端负责感受刺激的亮氨酸重复序列(leucine-rich repeat,LRRs)。当激活时,NLRP3的LRRs结构域可与NEK7直接结合,通过其N端的PYD/PYD同型相互作用招募含PYD和caspase招募域(caspase activation and recruitment,CARD)的ASC,相应地,ASC通过CARD/CARD相互作用招募含CARD域的caspase-1前体聚集[4]。同时,ASC通过PYDs寡聚形成螺旋簇状集合[5],即所谓的ASC斑点,催化无活性的caspase-1前体剪切p10、p20亚基[6],这两个亚基进一步结合构成有活性的四聚体形式,进而活化炎症因子IL-1β、IL-18,促进炎症免疫反应[7]。此外,活化的caspase-1还可诱导一种新型的被称为“细胞焦亡”的炎性细胞死亡,这种死亡方式依赖于一种称为gasdermin D(GSDMD)的蛋白的N端在细胞膜上形成纳米孔,导致细胞肿胀、释放炎性介质,进一步扩大炎症反应[8]。
1.2 NLRP3炎性小体的激活
无论是单独刺激NLRP3,还是过表达NLRP3以增强NLRP3对刺激的敏感性,均不足以诱导NLRP3炎性小体激活,说明它的激活机制较为复杂。研究发现,NLRP3炎性小体的激活需要两个步骤:第一步(启动),通过激活核转录因子NF-κB及其他转录因子,增强NLRP3、pro-IL-1β基因转录,提高其表达水平,并通过与转录无关的方式降低NLRP3炎性小体激活阈值;第二步(活化),当已启动的细胞受到激活剂刺激时NLRP3炎性小体就会被激活。与其他模式识别受体只能感受少数几种刺激不同,NLRP3可识别多种病原相关分子模式(pathogenassociated molecular patterns,PAMPs)和危险相关分 子 模 式(danger-associated molecular patterns,DAMPs),包括病毒RNA、尿酸单钠(monosodium urate,MSU)、细胞外ATP、离子载体尼日利亚菌素等[9]。此外,胞浆的脂多糖(lipopolysaccharide,LPS)还能引起caspase-11依赖性的非经典NLRP3炎性小体激活[10]。这些激活剂的相同之处在于它们都能诱导细胞应激,细胞应激被NLRP3感知,但NLRP3是如何感知细胞应激,以及哪些通路被诱导最终导致NLRP3炎性小体激活仍有待阐明。
NLRP3炎性小体的激活包含多个上游信号,包括活性氧(reactive oxygen species,ROS)上升、钾离子(K+)外排、溶酶体破裂、反式高尔基体网络结构(trans-Golgi network,TGN)解体、线粒体功能障碍等。例如,降低ROS的产生或使用ROS清除剂均能强有力地降低NLRP3炎性小体激活,但是有研究表明,抑制ROS并不直接影响NLRP3炎性小体的激活,而是负调控NLRP3炎性小体激活的启动步骤[11]。而大多数的NLRP3炎性小体激动剂都能引起K+外流[12],并且单独细胞外低钾便足以诱导NLRP3炎性小体激活,其机制为钾离子外流后导致NEK7与LRR结合,从而激活NLRP3炎性小体,但是有学者提出不同观点,Zhang等[13]认为其作用其实是影响静息膜电位,而且局部免疫调节剂咪喹莫特和相关分子CL097触发的NLRP3炎性小体激活与K+外排并无关[14]。关于溶酶体损伤导致的NLRP3炎性小体激活至今仍没有较明确的机制,目前认知仅限于通过胞吞作用引起溶酶体破裂,组织蛋白酶释放到胞质中,导致NLRP3炎性小体活化。但是,虽然溶酶体组织蛋白酶对NLRP3炎性小体激活十分重要,单独基因敲除并未见显著影响[15]。不同的NLRP3炎性小体激动剂均会引起TGN解体,之后通过带负电荷的磷脂酰肌醇-4-磷酸(phosphatidylinositol-4-phosphate,PtdIns4P)结合NLRP3,诱导衔接蛋白ASC聚合,激活下游信号传导级联[16],但为什么NLRP3炎性小体需要在分散的反式高尔基体网络上特异性组装,目前尚不清楚。因此,在关于NLRP3炎性小体激活信号的研究上仍存在许多争论。
2 钙离子信号与NLRP3炎性小体激活
离子流在NLRP3炎性小体激活过程中具有举足轻重的地位,而Ca2+作为调控细胞功能的一种重要离子流,其动态平衡障碍影响许多神经退行性疾病的发展[17]。在以NLRP3获得性突变为特征的冷热相关周期综合征(cryopyrin-associated periodic syndrome,CAPS)患者中,发现外周血单个核细胞内Ca2+升高后[2],钙离子在NLRP3炎性小体激活过程中发挥的作用开始被广泛研究。
2.1 Ca2+浓度控制方式
胞质Ca2+浓度通过Ca2+的流入和流出实现平衡,细胞质Ca2+内流有两种主要途径:(1)来自细胞内存储,如内质网(endoplasmic reticulum,ER)和线粒体;(2)来自胞外液。静息状态下,胞外液和ER腔内Ca2+水平维持在毫摩尔级,而细胞胞浆Ca2+水平较低,细胞质通过质膜钙ATP酶(plasma membrane Ca2+ATPase,PMCA)和肌质网膜钙ATP酶(smooth endoplasmic reticular Ca2+ATPase,SERCA)转运体的挤压将胞浆Ca2+浓度维持在100 nmol/L左右。另外,Na/Ca交换器(Na+/Ca2+exchanger,NCX)和Na/Ca/K交换器(Na+/Ca2+-K+exchanger,NCKX)是Ca2+的二级调节器,控制细胞内外钠钙交换[18]。
刺激条件下,Ca2+信号通路激活,质膜离子通道通过电压变化或与配体结合触发,造成细胞内外钙离子流动变化。胞浆Ca2+激增激活Ca2+结合蛋白和各种细胞活动。多种Ca2+可渗透通道参与细胞外Ca2+的内流:(1)电压门控Ca2+通道(voltagegated Ca2+channel,VOCs):膜去极化时激活,是电兴奋细胞中最主要的Ca2+流入方式;(2)受体门控通道(receptor-operated Ca2+entry,ROCs):当Ca2+激动剂结合到ROC,导致磷脂酶C(phospholipase C,PLC)信号通路激活,ER Ca2+释放;(3)配体门控的瞬时受体电位通道(transient receptor potential,TRP):是非兴奋性细胞主要的Ca2+流入方式,在膜和细胞器膜上多有分布,按氨基酸序列分为7个亚科:TRPC、TRPV、TRPA、TRPM、TRPML、TRPP和TRPN,并且能与Ca2+释放激活的Ca2+通道(Ca2+release-activated Ca2+,CRAC)相互作用,控制Ca2+信号;(4)存储操控通道(store-operated Ca2+entry,SOCs),如CRAC:当内质网Ca2+储存耗尽时,基质相互作用分子(stromal interaction molecule,STIM)与质膜中的Orai通道相作用,激活钙池调控钙离子通道(store-operated Ca2+entry,SOCE),引起细胞外Ca2+进入,以维持细胞内外钙离子的动态平衡[19],Ca2+可渗透通道如图1所示。
2.2 Ca2+信号在NLRP3炎性小体激活中的作用
早期研究主要是根据钙离子螯合剂BAPTAAM抑制IL-1β分泌的能力将Ca2+流动与NLRP3炎性小体激活联系起来的。如Chu等[20]发现BAPTA-AM可 抑 制NLRP3炎性小体激活和IL-1β分泌。接着,细胞外Ca2+被证明可单独刺激依赖于NLRP3炎性小体的IL-1β分泌。进一步研究发现,细胞外Ca2+或其他NLRP3炎性小体的激动剂可以通过钙传感受体(calcium-sensing receptor,CASR)和PLC之间的相互作用触发细胞内Ca2+信号级联反应,继而影响NLRP3[21]。Lee等[22]提出PLC介导的磷脂酰肌醇4,5-二磷酸(phosphatidylinositol 4,5-bisphosphate,PIP2)水解产物肌醇1,4,5-三磷酸(inositol 1,4,5-triphosphate,IP3)激活ER上的IP3受体(1,4,5-trisphosphate receptor,IP3R),进而触发ER Ca2+释放和NLRP3炎性小体激活。作者还发现,使用PLC的抑制剂抑制了多种刺激诱导的IL-1β分泌,而PLC的直接激活则可在没有任何其他刺激的情况下诱导NLRP3炎性小体激活。同理,敲除IP3R或药理学抑制其表达均可减弱Ca2+流动和NLRP3炎性小体激活。
此后,对其他多种不同结构激活剂刺激NLRP3炎性小体激活的研究进一步证实了Ca2+的作用。研究发现,脑心肌炎病毒(EMCV)通过刺激细胞内存储Ca2+释放到细胞质中进而激活NLRP3炎性小体[23]。登革热2型病毒及其非结构蛋白2A和2B激活NLRP3炎性小体与释放ER Ca2+有关[24]。ATP诱导的IL-1β释放需要细胞外Ca2+内流和ER介导的Ca2+释放[25]。紫外线诱导的NLRP3炎性小体激活需要升高的细胞内Ca2+[26]。胆固醇依赖性溶胞素激活NLRP3炎性小体需要Ca2+内流,而Ca2+内流对于植物凝集素激活NLRP3炎性小体也是必需的[27]。这表明细胞内Ca2+信号在NLRP3炎性小体激活过程中扮演着至关重要的角色。
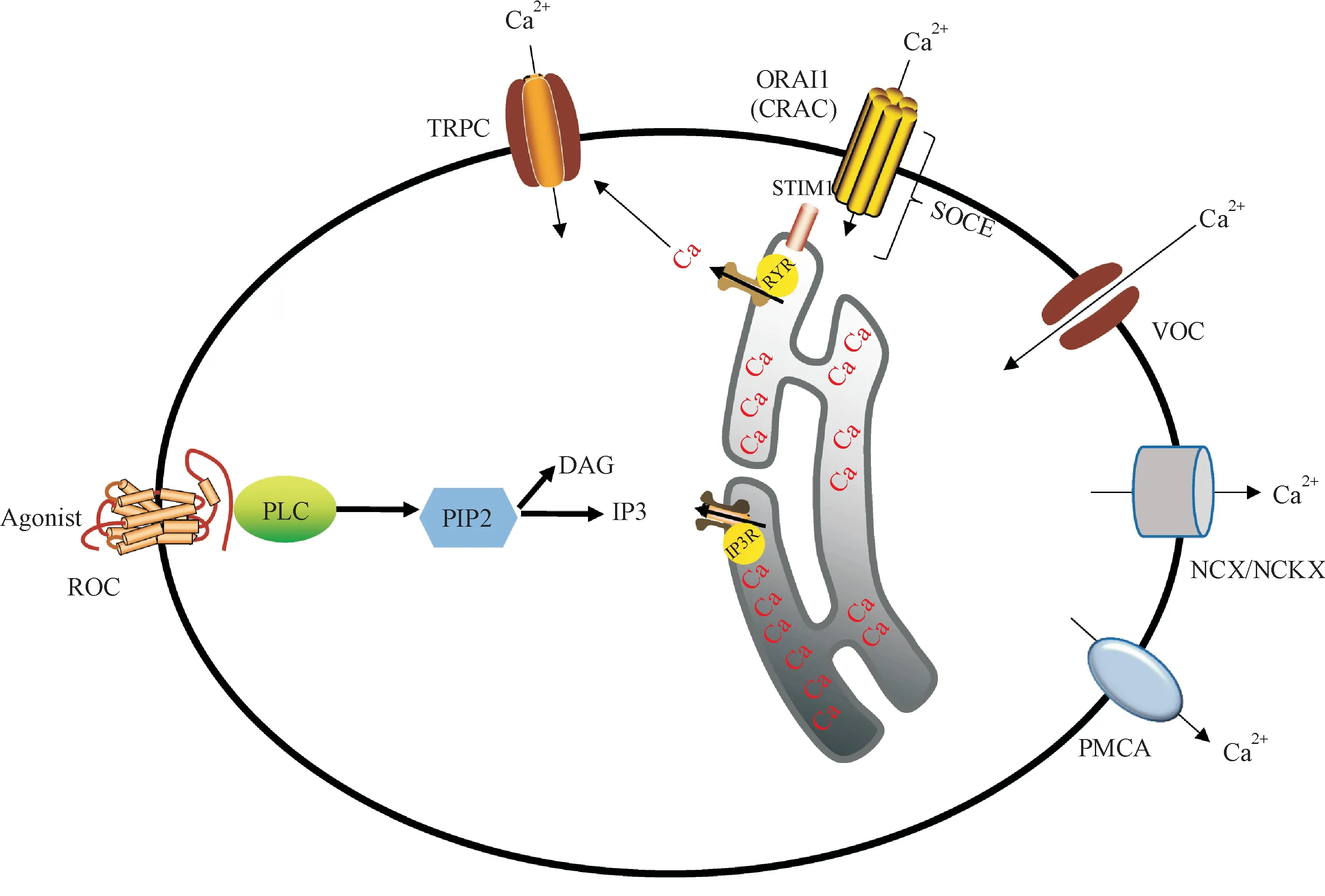
Figure 1 Ca2+regulation channel in cell
但是,针对Ca2+在NLRP3炎性小体激活中的作用,Katsnelson等[28]提出了不同看法,他们研究了NLRP3炎性小体激动剂刺激下,胞浆内钙离子上升对小鼠原代树突细胞、原代巨噬细胞NLRP3炎性小体激活情况和下游炎症信号反应的影响。首先,在内源性ATP门控P2X7受体通道、外源性离子载体尼日利亚菌素或促溶酶体LLME激活NLRP3炎性小体过程中,胞浆Ca2+增加并不是必要条件。其次,在小鼠树突状细胞中,与Ca2+流动相关的G蛋白偶联受体的激动剂对NLRP3炎性小体信号激活的作用被K+外排激动剂逆转。并且,虽然BAPTA-AM和2-APB强烈抑制了尼日利亚菌素诱导的NLRP3炎性小体信号传导,但其作用机制与Ca2+稳态无关[29]。因此,Ca2+在NLRP3炎性小体激活过程中发挥的作用仍需要进一步探索。
2.3 NLRP3炎性小体激活过程中Ca2+信号的变化
如上所述,在NLRP3炎性小体激活过程中常伴随着Ca2+的升高,但目前一个突出问题是NLRP3炎性小体激活过程中增加的细胞内Ca2+来源和机制。研究表明,在ATP/尼日利亚菌素刺激下,从细胞外进入和细胞内存储池中释放的Ca2+均有助于NLRP3炎性小体的活化[2]。
2.3.1 胞浆膜钙离子通道开放 在NLRP3炎性小体激动剂刺激下,胞浆膜上的钙离子通道发生变化,影响胞浆内钙离子浓度。钙内流是NLRP3炎性小体激活过程中的近端步骤,而NLRP3炎性小体的不同激活剂可能使用不同的质膜离子通道介导钙内流:(1)激活电压门控Ca2+通道(VOCs):质膜极化可调节Ca2+信号,在NLRP3激动剂诱导的细胞外Ca2+进入过程中,K+外流抵消了膜去极化作用,从而促进Ca2+进一步内流[13];(2)激活受体门控通道(ROCs):细胞外ATP与嘌呤能受体P2X结合后,导致质膜中的阳离子渗透通道开放,诱导胞质钙增加[30];细胞外ADP通过P2Y1受体介导钙信号,增加Ca2+流动,并以细胞外信号调节激酶5(extracellular signal-regulated kinase 5,ERK5)依赖性方式激活NLRP3炎性小体[31];增加的[Ca2+]ex通过G蛋白介导的CASR信号通路触发单核细胞和巨噬细胞大胞饮[32];(3)激活配体门控的瞬时受体电位通道(TRP):在LPS启动NLRP3过程伴随TRP通道依赖性Ca2+信号转导,LPS诱导细胞产生第二信使甘油二酯(diacylglycerol,DAG),DAG以Toll样受体4(Toll-like receptor 4,TLR4)依赖的方式直接激活TRPC6,TRPC6介导的Ca2+流入反过来激活肌球蛋白轻链激酶(myosin light chain kinase,MYLK),导致LPS/TLR4介导的NF-κB激活[33]。而在NLRP3激活过程中,晶体或脂质体刺激诱导ROS爆发,继而产生代谢物ADP-核糖,通过TRPM2通 道诱 导ROS依 赖的 钙内 流 和IL-1β释放[34];并且TRPM2同一家族的其他成员(TRPM7、TRPV2、TRPA1和TRPV1)也与炎性小体激活有关[35-36]。(4)激活存储操控通道(SOCs):当内质网Ca2+储存耗尽时,激活SOCE,引起细胞外Ca2+进入,而抑制SOCE的主要组分STIM1、SERCA2均显著阻碍了NLRP3炎性小体的组装[37]。
2.3.2 细胞存储的钙离子释放 细胞存储的钙离子也会释放到胞浆中。内质网作为细胞内钙离子的主要存储库,其钙释放对维持钙稳态是极其重要的,并且阻断内质网上钙释放通道影响NLRP3炎性小体激活。使用药理方法抑制内质网主要的钙离子释放通道IP3R可阻断多种激活剂诱导的NLRP3炎性小体激活。通过阻断其上游的PLC产生IP3同样能抑制NLRP3炎性小体激活。此外,其他内质网钙离子释放通道,如兰尼碱受体(ryanodine receptor,RyR)通道开放也能从内质网腔释放钙离子[38]。溶酶体作为酸性Ca2+存储体,具有一系列Ca2+可渗透通道,以允许Ca2+释放[39],在MSU诱导的NLRP3炎性小体激活模型中,往往伴随着溶酶体膜破坏,溶酶体内Ca2+流出[40]。
2.4 Ca2+信号促进NLRP3炎性小体激活的机制
2.4.1 Ca2+与cAMP平衡 尽管关于Ca2+的流动与NLRP3炎性小体激活的关系已有大量研究,但Ca2+流动导致NLRP3炎性小体激活的分子机制仍存在许多空白。研究发现Ca2+可促进LPS刺激的巨噬细胞游离裂解液中自发的NLRP3-ASC结合,提示Ca2+可直接调节NLRP3炎性小体的激活。但是研究并没有阐述Ca2+如何促进复合物形成,以及Ca2+的直接靶点究竟是什么。另外,研究还揭示了环磷酸腺苷(cyclic adenosine monophosphate,cAMP)生成与NLRP3激活的关系,cAMP与NLRP3直接结合抑制炎性小体组装,而CASR激活导致cAMP下调,释放被抑制的NLRP3。在CAPS患者体内,由于存在NLRP3功能获得性突变,cAMP与NLRP3的亲和力明显低于正常,并且通过增加cAMP能降低CAPS患者外周血单核细胞中的IL-1β。该研究有力地表明细胞内NLRP3炎性小体的状态取决于Ca2+与cAMP的平衡[22]。
之后的研究证实并对此发现进行了补充。一些代谢物,如醋酸盐可通过PLC-IP3途径减少Ca2+流动,并伴随着cAMP增加,之后活化PKA,通过自噬或蛋白酶体途径泛素化降解NLRP3,从而抑制炎性小体激活[41]。Ca2+浓度的升高也会激活下游JNK信号通路以及诱导后续的ASC接头蛋白低聚。在乙醇诱导的神经元细胞中,乙醇通过N-甲基-D-天冬氨酸受体(NMDA receptor,NMDAR)诱导细胞内Ca2+增加,激活钙调蛋白依赖的蛋白激酶Ⅱ(calmodulin dependent kinase II,CAMKⅡ),之后通过激活JNK1促进NLRP3炎性小体组装和活化[42]。Chen等[43]首次发现细胞六型分泌系统效应蛋白EvpP能通过抑制Ca2+依赖性MAPK-JNK通路,影响ASC低聚,阻断NLRP3炎性小体激活。最新研究表明,细菌感染通过Ca2+结合蛋白TBC1结构域家族成员9(Ca2+-binding protein TBC1 domain family member 9,TBC1D9)增加Ca2+水平,激活TBK1,从而促进自噬[44],而自噬与NLRP3炎性小体的激活密切相关[45]。
2.4.2 线粒体损伤Ca2+流动激活NLRP3炎性小体的另一重要机制是通过诱导线粒体损伤。Ca2+通过线粒体内膜中的线粒体钙单向转运体(mitochondrial calcium uniporter,MCU)和线粒体外膜中的电压依赖性阴离子选择通道(voltage dependent anion selective channel,VDAC)流入线粒体,导致线粒体钙超载,线粒体活性氧(mitochondrial reactive oxygen species,mROS)增加,线粒体膜电位下降,通透性转换孔开放,以及释放线粒体DNA(mitochondrial DNA,mtDNA)和心磷脂。研究人员证实,在ATP刺激过程中,Ca2+流动的关键作用就是诱导线粒体损伤,接着产生的mtDNA和心磷脂,经氧化直接结合NLRP3[2]。其中细胞质mtDNA与NLRP3共定位并增强IL-1β的分泌,而氧化的mtDNA进一步诱导IL-1β产生[46]。而线粒体膜电位下降和通透性转换孔开放,释放细胞色素c,细胞色素c通过抑制呼吸链复合物Ⅲ来促进ROS产生,ROS一方面进一步激活IP3R,另一方面通过与硫氧还原蛋白(thioredoxin-interacting protein,TXNIP)结合激活NLRP3[47]。
Ca2+流动通过影响线粒体影响NLRP3炎性小体激活的机制得到了广泛认可。首先,从结构上来说,内质网和线粒体的空间排列促进了内质网和线粒体之间的联系,这对于Ca2+从内质网转移到线粒体和NLRP3炎性小体激活都是至关重要的[48]。其次,研究证实,多种NLRP3炎性小体激动剂都能使线粒体钙离子超载,而减少线粒体钙离子能降低mROS水平,减少mtDNA产生,降低炎性水平[2]。Li等[49]使用高浓度葡萄糖诱导血管平滑肌细胞(VSMCs)糖尿病模型,发现该造模过程激活Ca2+信号通路,导致线粒体损伤,释放mtDNA至胞浆,影响糖尿病小鼠NLRP3炎性小体激活和VSMCs重塑。并且,影响线粒体钙离子进入通道影响炎症反应已得到实验支持。在铜绿假单胞菌感染的原代人气道上皮细胞模型中,由于线粒体Ca2+超载导致的线粒体失调促进NLRP3炎性小体激活[50];药理学抑制MCU显著改善体内外炎症反应[51]。最后,Ca2+本身就控制着线粒体动力学的许多方面。例如,Ca2+影响线粒体的能动性,而微管驱动的线粒体与内质网的结合是NLRP3炎性小体复合物组装的必要条件[48];Ca2+调控线粒体融合和分裂的相关过程,而线粒体融合的关键调控因子Mitofusin 2已被证明在RNA病毒感染模型中影响NLRP3炎性小体活化水平[52];在少突胶质细胞和阿尔茨海默病转基因小鼠中,下调线粒体分裂蛋白Drp1表达极大程度上降低了NLRP3炎性小体激活和炎性反应[53]。
2.4.3 干扰Ca2+敏感的酶 除此之外,激活或失活Ca2+敏感的酶,也能影响NLRP3炎性小体的激活。经典的NLRP3炎性小体激活剂刺激导致Ca2+流动,增加钙蛋白酶活力,活化的钙蛋白酶释放被细胞骨架隔离的caspase-1,以调节NLRP3炎性小体活化,而使用不同机制的钙蛋白酶抑制剂均能降低IL-1β水平,反之,敲除内源性的抑制剂钙蛋白酶抑制蛋白(CAST)增强IL-1β水平[13]。Ca2+信号调节NLRP3炎性小体激活的机制如图2所示。
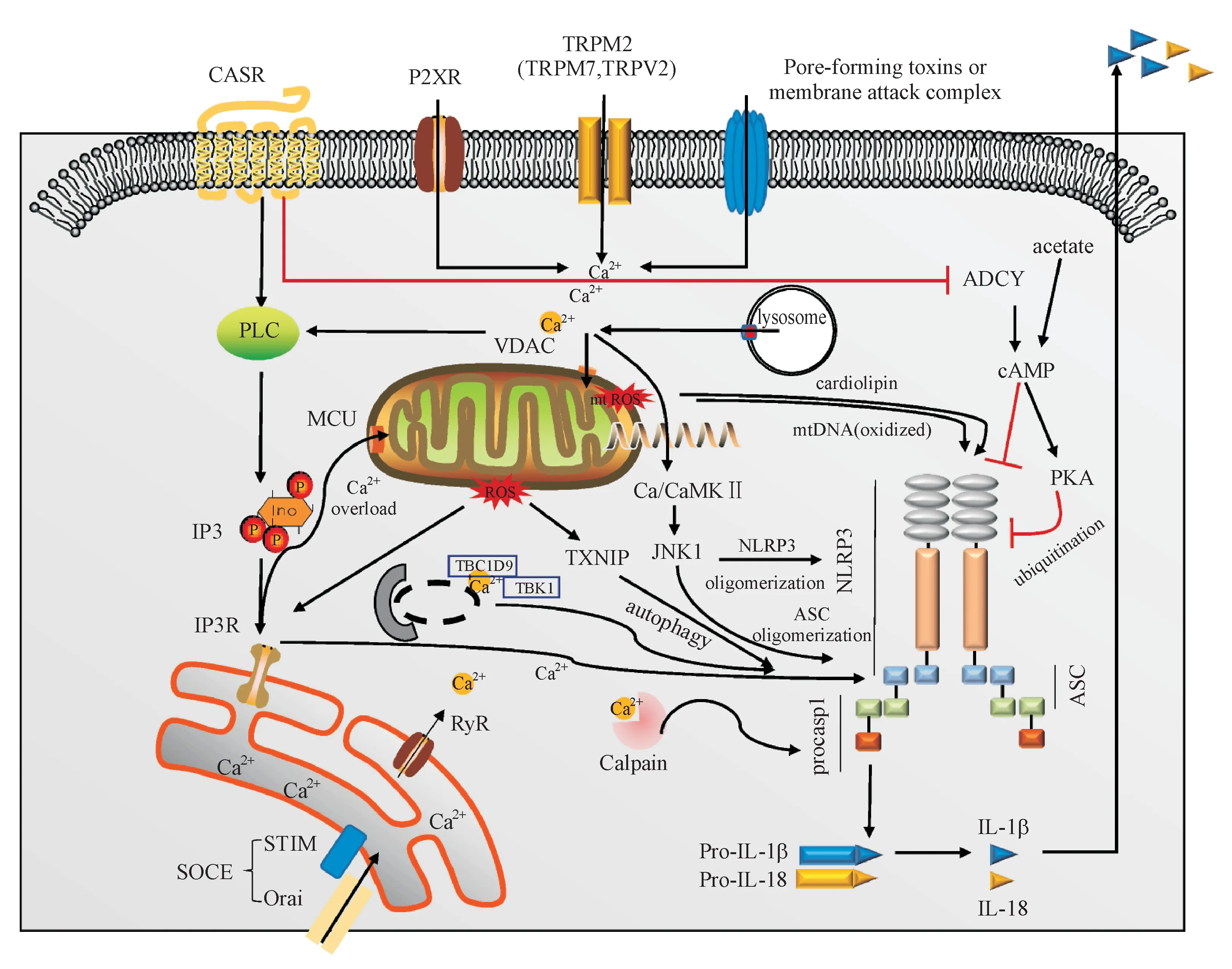
Figure 2 Regulation of NLRP3 inflammasome activation by Ca2+signaling
3 调控Ca2+的相关药物及靶点研究现状
3.1 镇痛
在LPS诱导的炎性疼痛小鼠模型中,Yin等[54]发现芍药苷可通过抑制TRPV1激活,降低细胞内Ca2+浓度,抑制PKC活性,最终导致减少NLRP3炎性小体激活及caspase-11介导的非经典细胞焦亡,起到镇痛作用。
3.2 心力衰竭
最近,新型口服降糖药物钠/葡萄糖共转运体2(SGLT2)抑制剂恩格列净和卡格列净,已被证实与2型糖尿病患者心血管死亡和心力衰竭住院率降低相关[55-56]。恩格列净可减轻2个无高血糖和糖尿病的心衰模型(射血分数保持的心力衰竭HFpEF和射血分数降低的心力衰竭HFrEF)的心功能障碍。这些有益的心脏效应与心肌NLRP3炎性小体通路和下游细胞因子信号通路的激活减少有关。此外,研究发现恩格列净以Ca2+依赖的方式减弱心肌细胞NLRP3炎性小体启动,这表明钙离子稳态在恩格列净抗心衰过程中发挥重要作用[57]。
3.3 其他
激活配体门控的瞬时受体电位通道是药物研发的重要靶点,其中TRPV4是一种广泛表达的Ca2+通道[58]。TRPV4通过调节Ca2+进入调节各种生理过程相关的细胞信号传导,是治疗人类疾病的一个重要靶点。TRPV4通过增加脑出血后的胞浆内Ca2+触发的炎症反应导致了小鼠神经元死亡[59];孟鲁司特通过调节TRPV4通道抑制出血性膀胱炎大鼠模型损伤[60]。事实上,针对TRPV4拮抗剂的研究在过去的十年中蓬勃发展。据报道,超过10种独特的化学类型可抑制该离子通道[61],第1个TRPV4拮抗剂GSK2798745已经进入临床试验阶段[62-63]。
4 结语和展望
在过去的十年中,已经证实了Ca2+在NLRP3炎性小体激活中的关键作用,但关于Ca2+与NLRP3炎性小体激活的关系仍存在许多问题有待阐明。本文综述了Ca2+与NLRP3炎性小体激活的关系,重点梳理了已有的关于Ca2+流动激活NLRP3炎性小体的机制,包括直接结合NLRP3、通过CAMKIIJNK影响ASC低聚、造成线粒体钙超载引发线粒体失稳和影响Ca2+敏感的酶。多种形式的细胞应激,包括质膜损伤、溶酶体损伤和离子失衡,都可能影响Ca2+流动来激活NLRP3炎性小体。进一步研究钙离子并且了解NLRP3炎性小体激活和调控的详细机制将进一步促进对炎症的理解,并能为治疗NLRP3炎性小体驱动的炎症性疾病提供新思路。
