儿童共济失调毛细血管扩张症4例病例报告
2021-10-22杨家鑫
杨家鑫 蒋 莉
1 病例资料
例1:男,8岁6个月,因“步态不稳5年余,发现头颈右偏伴抖动、痰中带血4 d”入重庆医科大学附属儿童医院(我院)神经内科。患儿自2~3岁起发现步态不稳,呈醉酒步态,伴持物震颤、语言发育较缓慢,左手拇指内收、手指轻微爪形现象,逐渐加重。于当地医院诊断为“发育迟缓”,未予以特殊治疗。7岁时步态不稳及言语不清进行性加重,头颅MR提示小脑萎缩(图1)。8岁左右出现躯干及四肢的不自主抖动伴肌张力障碍,头向右偏,紧张时明显,入睡后头位可恢复正常。
G3P2,足月顺产,无产伤及窒息史。1岁6月时可独立行走,但行走不稳,易跌倒。现上小学二年级,成绩中等。每月1~2次呼吸道感染。G1P1,患儿姐姐14岁,体健,G2孕4月自然流产。父亲为乙肝携带者,母亲体健,否认近亲婚配,否认3代内有类似症状者。
神经系统体格检查:神志清楚,语速缓慢,头向右偏,未见明显球结膜充血扩张,无眼球震颤,眼球活动正常。腹部可见一块3.5 cm×3.0 cm咖啡斑,左手拇指内收呈爪形,四肢肌力正常、肌张力障碍明显。双侧腱反射减弱,巴氏征阴性。指鼻试验及跟膝胫试验阳性。AFP 117(正常值<6)ng·mL-1,IgG及IgM未见异常,IgA低于正常值(表1),头颅MR示小脑萎缩。

图1 例1的头颅MR表现
例2:女,8岁5个月,因“发现步态不稳6年余,加重1年”入我院。2岁左右出现步态不稳,易跌倒,后逐渐出现身体晃动。7岁站立时有身体晃动,不能维持直线行走,需扶着上下楼梯,伴语速减慢、吐词不清、流涎。
G4P1,因胎膜早破行剖宫产,前3胎人工流产。现上小学二年级,成绩较差,韦氏智力测试:IQ 68分。平素易反复呼吸道感染。父母均体健,否认近亲婚配和类似疾病家族史。
神经系统体格检查:神志清楚,反应较迟钝。双眼轻微水平眼震,双眼球结膜及颜面部皮肤有毛细血管扩张(图2),眼球活动迟钝。伸舌稍向右偏。言语含糊,语速慢,语调异常。书写不流畅,字大。有躯体震颤,指鼻试验、跟膝胫试验、轮替试验和闭目难立征阳性。四肢肌力和肌张力正常。双侧腱反射减弱,巴氏征阴性。AFP 99.3 ng·mL-1,IgG降低、IgM升高,IgA降低(表1),我院头颅MR示小脑萎缩。
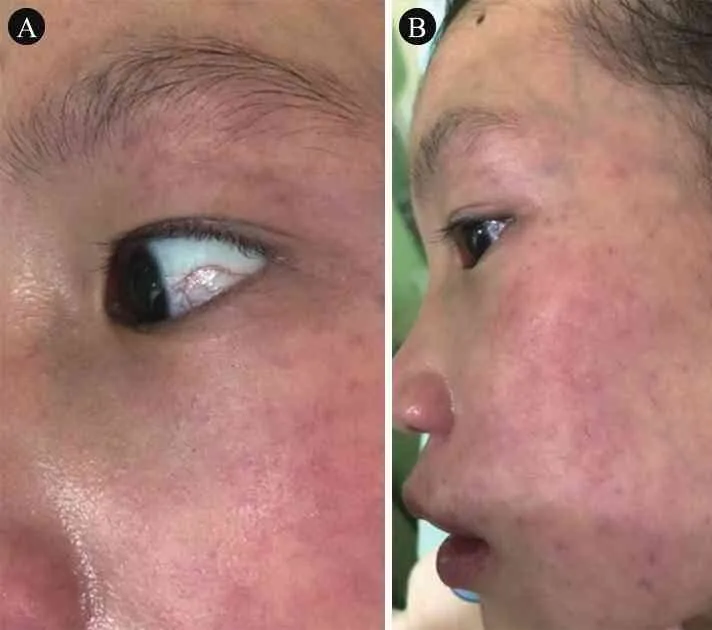
图2 例2球结膜及颜面部皮肤毛细血管扩张
例3:男,7岁11个月,因“运动发育落后7年,步态不稳3年余,进行性加重半年余”入我院。患儿自幼运动能力落后,走路跑步较同龄儿发育缓慢。4岁时加重,有轻度步态不稳,呈醉酒样步态,头部不自主运动,躯体晃动伴流涎,可跑、并足跳,可简单的语言交流。7岁时运动能力进行性低下,步态不稳明显,不能走直线,不能独站,可扶走,语言减少、吐词不清、对答缓慢,只可计算5以内加减法,记忆力变差。7岁半时头部、四肢不自主运动较前增多。
G2P1,G1孕5月时因“头部畸形”行引产,G3P2为患儿5岁妹妹,体健。父母体健,否认近亲婚配和有类似疾病家族史。
神经系统体格检查:神志清楚,可理解和执行指令。可见头部不自主运动。双眼水平眼震,眼球活动无异常。吟诗样语言,吐词不清。书写不流畅,字体大小异常。躯体震颤及持物震颤明显,指鼻试验、跟膝胫试验、轮替试验、闭目难立征阳性。头颅MR示小脑萎缩。
例4:男,6岁2个月,因“发现步态不稳、言语不连续4年余”入我院。患儿自2岁起出现步态不稳,呈蹒跚步态,易摔倒,站立时身体摇晃,持物震颤明显,说话语句不连续,说话时口角稍向左外斜,易流涎,外院头颅MR未见异常。
G1P1,因“羊水污染”剖宫产。1岁多时开始行走,说单字或短句,现上幼儿园,学习能力较同龄儿稍差。父母体健,非近亲婚配,否认类似疾病家族史。
神经系统体格检查:神志清楚,双侧球结膜轻微充血,颈背部及腰部皮肤有毛细血管扩张,口咽部不自主运动,语言欠流畅,吐词尚清楚。躯体震颤较明显,指鼻试验、轮替试验可疑阳性,闭目难立征阳性;跟膝胫试验阴性。四肢肌力、肌张力正常。双侧腱反射亢进,巴氏征阴性。AFP 1.83 ng·mL-1,免疫功能未见异常(表1),在我院行头颅MR未见异常。
随访:4例均随访至2021年6月,例1和例3已不能独立行走。
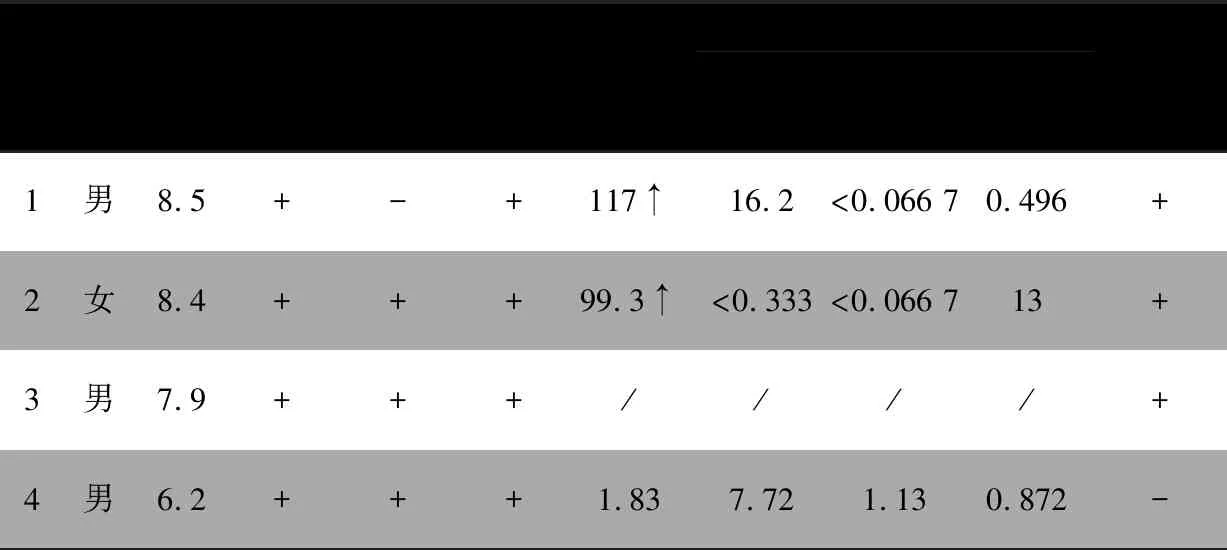
表1 4例共济失调毛细血管扩张症患儿的临床资料及实验室检查结果
2 基因检测
经我院医学伦理审核及监护人知情同意,采集患儿及父母外周血,由智因东方、迈基诺、嘉检等第三方检验公司完成基因测序。4例患儿均接受了二代DNA测序及Sanger测序验证,表2显示,例1、2和4为复合杂合突变,例3为纯合突变,共检测到共济失调毛细血管扩张症基因(ATM)基因7种突变位点,3个移码突变,1个剪切位点突变,2个错义突变,1个大片段基因缺失;其中c.974_975del、c.5706_c.5707insA、lossl (exon62-63)在千人数据库、dbSNP数据库及hapmap数据库中未见报道,经蛋白质功能预测软件Polyphen-2 和Mutation Taster预测均为有害。Sanger测序显示,4例患儿父母均为基因突变携带者,根据ACMG(American College of Medical Genetics)[1]指南,7个基因突变均为“致病性变异/可能致病”。

表2 4例患儿ATM基因变异情况
3 讨论
共济失调毛细血管扩张症(AT)是一种累及多个系统的常染色体隐性遗传原发性免疫缺陷疾病[2]。由复合杂合突变或纯合突变所致,以复合杂合突变为主。典型表现为进行性的小脑共济失调、球结膜毛细血管扩张、免疫缺陷、反复的副鼻窦炎和肺部感染。
神经系统异常是AT患者的主要临床表现,共济失调常是首发症状。Levy等[3]总结的AT病例资料显示,96%(344/357)有小脑共济失调,本文4例均有小脑共济失调。大多患儿学走路时即出现步态异常,4~5岁时明显,逐渐加重[4],本文3例2岁左右即出现共济失调,提示该表现为AT早期识别的要点。既往报道,构音障碍(80%)也是AT的典型表现[5],本文4例均有此表现。Woods和Taylor[6]首次报告,79%(55/70)的患者有肌张力障碍,包括面部、四肢、舌和躯干肌张力障碍,但无眼睑痉挛,在疾病后期,头部痉挛可能代表颈部肌张力障碍。本文例1有明显肌张力障碍及头颈右侧痉挛,与上述基本相符,表明肌张力障碍也可能是AT的一个主要特征。本文3例出现球结膜毛细血管扩张,证明其为AT的典型临床表现,其原因可能与ATM突变和HIF-1蛋白表达增加有关,HIF-1过表达使HIF-1的靶基因VEGF表达增加,导致血管生成[7]。毛细血管扩张常在5~8岁时发生,有时更晚或者不发生[4]。因此,在长期反复共济失调的患者中若出现球结膜充血等要注意鉴别AT。此外,AT患者小脑皮质变性会导致异常的眼球运动和视觉异常,包括眼球运动性失用、周期性眼震、凝视性眼震及斜视,本文例2、例3出现眼震。
本文例1和例2有反复呼吸道感染病史,提示该病患者易出现免疫缺陷,有学者认为ATM缺失会使炎性小体复合体氧化抑制,导致先天免疫功能下降[8],因此一般建议完善免疫功能相关检测。低水平IgA被报道为AT患者淋巴癌发展的危险因素[9],故本文IgA下降的2例患儿应长期随访。AFP是AT患者的诊断标志物,通常由胚胎卵黄囊和胎儿肝细胞产生,主要在发育的前3个月表达,随后几年减少。在AT患者中95%以上出现AFP升高[10,11]。本文3例行AFP检测的患儿中,2例升高,提示临床中对于AFP升高的共济失调患者需要高度警惕AT。本文3例在2岁左右出现运动功能障碍,其中2例伴反复呼吸道感染,但确诊年龄却在6岁半及8岁半,提示若发现患儿自幼出现运动障碍且易反复感染时,应警惕该病,完善免疫学相关检查及AFP的筛查。若患儿还出现毛细血管扩张,特别是年龄为5~8岁,则应该高度怀疑AT,建议完善基因检测。
本文ATM基因突变3例为复合杂合突变,1例为纯合突变,大部分为移码突变,符合既往文献报道的ATM突变特征[12]。例1的c.974_975del、c.7878_7882del,例2 c.5706_c.5707insA均为移码突变,可以导致蛋白质的截断突变引起酶的表达障碍,从而产生强致病性,其中c.974_975del(p.I326Rfs*3)尚未有文献报道,p.I326Rfs*3突变在ATM基因中产生过早的翻译终止信号,根据预测软件其会导致蛋白质产物缺失或破坏从而致病。例2的c.1899-1G>A为剪切位点的突变,预测软件表明该变异对RNA剪接有影响,可能会破坏共有剪接位点,从而导致蛋白质功能丧失,虽然该变异根据ACMG分类为可能致病,但结合该患儿典型的AT临床表现,判定为致病性变异。例3 c.6679C>T及例4的c.6115G>A均为错义突变,c.6679C>T (p.R2227C)已经在多个临床病例中被报道致病[13];例4 c.6115G>A(p.Glu2039Lys)序列在ATM蛋白的密码子2 039处用赖氨酸替换谷氨酸,有研究表明这种错义突变将导致ATM激酶活性降低[14],软件预测蛋白质功能将受到影响,且患儿的ATM基因exon 62-63处有428 bp的大片段基因缺失,结合患儿临床表现与基因突变高度相符,判定为致病性变异。
作为一种罕见的常染色体隐性遗传疾病,基因检测是诊断AT的金标准,本研究报道了4例儿童期起病AT患儿,发现了7个ATM基因突变位点,其中c.5706_c.5707insA、c.974_975del、lossl (exon 62-63)尚无文献报道。临床出现进行性小脑共济失调、球结膜毛细血管扩张、构音障碍、反复的呼吸道感染等累及多系统表现时应考虑此病;该病尚无特效治疗,尽早干预可避免反复呼吸道感染和放射线暴露。对症治疗与康复训练有助于改善神经功能,并能通过遗传咨询、准确的产前诊断降低发病率。
