ABCC9新发突变致多毛性骨软骨发育不良综合征1例病例报告
2021-10-22戴杰民
赵 欣 戴杰民 袁 萍 程 敏 蒋 莉 钟 敏
1 病例资料
查体: 神清,反应欠佳,R 36次/min,P 115次/min,双侧瞳孔等大等圆,对光反射灵敏,体重 4.5 kg (Z分值:-1.74),身高52 cm (Z分值:-2.99),头围39 cm (Z分值:-0.11 ),胸围39 cm,腹围40 cm,发育及营养中等。前额及臀部毛多,头发浓密,鼻梁稍宽,嘴巴宽,嘴唇丰满,鼻梁低平,人中长,未见上眦赘皮(图1)。双肺呼吸音粗,可闻及少许粗湿啰音,未闻及哮鸣音,心脏听诊未闻及杂音。肝肋下2 cm,质软;脾肋下1.5 cm,质中。
辅助检查:血常规、肝肾功能、电解质、血氨和乳酸、心肌标志物和酶谱、性激素水平未见异常。心电图:T波改变,QT间期延长。心脏彩超:右房、右室增大,右室前壁及室间隔增厚,肺动脉增宽,肺动脉压力测值增高(重度);室间隔基部缺损(小,双向分流);动脉导管未闭(小,右向左分流为主);房间隔卵圆孔未闭(双向分流);三尖瓣中度返流,肺动脉瓣轻中度返流;左心功能测值未见异常。胸部CT平扫+增强(图2):肺动脉主干较同层主动脉层面宽,肺动脉高压可能,双肺多血改变,伴广泛炎症。四肢长骨X线片:两侧股骨及胫骨见薄层骨膜反应。头颅MR平扫:脑含水量多,脑白质髓鞘化延迟,左侧颞叶可疑出血灶。脑电图:睡眠期两侧前额-额区尖样慢波阵发性出现。腹部+肾上腺彩超:肝脾稍肿大。
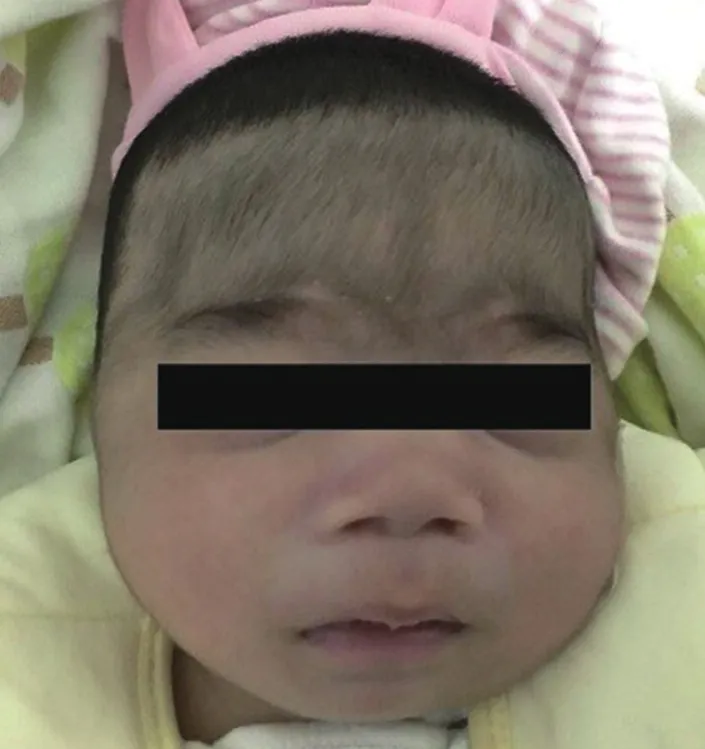
图1 患儿面容

图2 胸部CT
遗传学检查:染色体核型分析未见异常。经患儿父母知情同意后,抽取患儿及父母外周血各2 mL(北京智因东方转化医学研究中心)行全外显子测序。结果显示:患儿ABCC9基因的 27号外显子c.3460C>T(p.Arg1154Trp) (NM_005691)新发杂合突变,根据ACMG评级系致病突变,先证者及其家系成员表型及基因型的共分离,支持致病。该变异位点不同种属之间高度保守。此外,全外显子测序发现患儿有SCN9A(chr2)1c.536 (exon5)G>A,p.G179E(NM_002977)杂合突变,来自于没有惊厥等表型的母亲,不支持该位点致病。未发现和表型相关的其他致病位点。

本组病历资料、患儿照片等信息的使用得到其监护人知情同意,并签署知情同意书。本研究通过重庆医科大学附属儿童医院科研伦理审批(2018-64)。
2 讨论
CS(OMIM:239850)是一种表现为先天性多毛症、面部异常伴骨骼和心脏缺陷的常染色体显性遗传病,特征是先天性多毛症、新生儿巨体症、明显的骨软骨发育不良和心脏肥大[1]。多毛症导致浓密的头皮毛发延伸到前额,体毛普遍增多。相对特异的体征包括:大头畸形、面部粗大,鼻梁宽且低平,上眦赘皮,嘴巴宽,嘴唇丰满,人中长。约半数患儿出生时为巨大儿或有水肿。儿童期常表现为皮下脂肪少,颅骨增厚,胸腔狭窄,肋骨宽,椎体扁平或卵圆形,髋外翻,骨质减少,髓管增大,长骨干骺端变宽。约80%的病例有动脉导管未闭、心室肥大、肺动脉高压和心包积液等。运动发育异常通常表现为肌张力低,运动发育延迟。多数患儿有轻微的语言延迟,少数有学习困难或智力障碍[1, 2]。
以(Cantú Syndrome) OR (Cantu syndrome)或“多毛性骨软骨发育不良综合征”为关键词分别在中国期刊全文数据库(CNKI)、万方数据知识服务平台、PubMed和HGMD数据库检索,时间为建库至2019年11月30日。检索到34篇中英文病例报告,共87例CS,男35例(40.2%),女51例,1例未报告性别。文献中CS病例临床表现的数据不完整,主要有:羊水过多 (72.5%,29/40),出生超重(72.0%,36/50);多毛症和特殊面容(71.3%,62/87),头围偏大(75.0%,42/56);其他表现主要有发育迟缓、牙龈增生、反复上呼吸道感染、关节过度伸展等;心脏超声常见异常包括心肌病(59.1%,26/44)、动脉导管未闭(65.2%,30/46)和肺动脉高压(50.0%,12/24);X线片可见肋骨宽大(79.4%,27/34)、厚颅盖(33.3%,14/42)和干骺端扩大(45.5%,20/44)。
CS自1982年首次报道[2]以来,2005年首次发现其发病与染色体核型异常(1p36缺失)有关[3]。2012年发现致病基因ABCC9突变[4,5 ],2013年报道KCNJ8基因突变也可致本病[6, 7]。文献检索到的87例CS患儿中,有确切遗传学病因的占64.4%(56例)。其中,除2例KCNJ8突变和3例染色体核型异常外,均为ABCC9突变致病(92.9%,52例),c.3460C>T, p.Arg1154Trp (NM_020297.2, NM_005691.3)和c.3461G>A,p.Arg1154Gln (NM_020297.2)是最常见的突变类型[8~11]。
ABCC9(曾名SUR2)位于人类染色体12p12.1,编码1 549个氨基酸的跨膜蛋白。ABCC9蛋白是ATP敏感钾通道复合体组分之一,由2部分组成:①4个亚基[KCNJ8(又名Kir6.1)或KCNJ11(又名Kir6.2)]组成的钾通道;②4个调节功能的ABCC9亚基。ABCC9有2种组织特异性剪接体,SUR2A主要表达于心脏和肌肉,SUR2B表达于血管平滑肌和毛囊[4~6,10],其差别在于最后1个外显子不同。SUR2A特有的外显子杂合突变与扩张性心肌病和房颤有关。所有CS病例中发现的突变点都位于上游外显子,故可影响这两种剪接变体。因此,CS患儿可同时有心血管和毛发异常表现[4~6,10]。KCNJ8和ABCC9的编码蛋白共同构成上述ATP敏感钾通道复合体,因此KCNJ8突变致CS也得到证实[6,7]。
由此可见,CS是以心血管、毛囊、骨骼等多系统受累的罕见遗传病,ABCC9突变是其最主要的病因,以多毛为主的特异性外貌。对孕期或出生后早期疑诊病例应尽早评估,积极治疗心血管并发症,为其后期手术争取最佳时机。基因检测可明确遗传病因,以对家系成员生育提供遗传咨询。
