不同煎煮时间下10批三果汤散的HPLC指纹图谱研究
2021-10-20张艳娇廖振中林艾和朱培芳
张艳娇 廖振中 林艾和 李 蓉 黄 宽 范 源,3* 朱培芳
1.云南中医药大学中药学院,云南 昆明 650500;2.云南迪庆藏族自治州藏医院,云南 迪庆 674499;3.云南中医药大学第二附属医院,云南 昆明 650216
三果汤散由余甘子(PhyllanthusemblicaLinn.)、诃子(TerminaliachebulaRetz.)和毛诃子[Terminaliabellirica(Gaertn.) Roxb.]三味药材磨成细粉,按比例为1∶1∶1组成,是藏医最常见的基础方,有研究表明[1]三种药物比例相等时,存在正相互作用,又称“哲布松汤”,在印度阿育吠陀医学中应用也较为广泛。三果汤散具有抗氧化[2]、抗红细胞增多症[3]、抗糖尿病[4]的作用;最新研究[6]表明三果汤具有调节肠道微生物[5]、治疗口腔疾病如牙周炎、抗微生物感染[7]、影响肿瘤血管生成[8]等作用。其中,酚酸鞣质类成分是三果汤的主要化学成分,较多研究者对其单一成分如没食子酸(Galiic acid,GA)进行质量控制研究,或者分别测定余甘子、诃子、毛诃子三种药物中GA的含量[9-11]。李斌等[12]对三果汤中的GA、柯里拉京(Corilagin)、鞣花酸(Ellagic acid,EA)进行含量测定,张海伟等[13]采用HPLC法对不同批次藏药三果汤中主要酚酸类成分GA、EA进行含量测定。目前相关报道对三果汤散质量控制研究都不全面,测定时因其特征成分不统一,专属性差,难以实现质量控制标准化[14]。故增加指标性成分检测及指纹图谱的建立可更好地为三果汤的质量控制研究提供依据。
传统中药的煎剂具有悠久的历史,现代中医临床仍应用广泛[15]。中药所含的化学成分会随煎煮时间不同发生变化,根据疾病的不同,中药的煎煮时间的确立具有重要意义。指纹图谱分析是近代药物尤其是组方中药用得最普遍的一种质控研究方法,其是评价中药质量一致性、真实性、产品稳定性的一种可行性模式分析[16]。本实验采用HPLC法指纹图谱结合化学模式分析不同煎煮时间下的10批三果汤散的稳定性,以期为三果汤散的质量控制研究及临床应用提供参考。
1 实验材料
1.1 仪器 KDM调温电热套,常州国华电器有限公司;EYEL4型旋转蒸发仪,上海爱朗仪器有限公司;YL-080ST型超声清洗机,深圳市语路清洗设备有限公司;FA1004N型万分之一电子分析天平,上海菁海仪器有限公司;Agilent 1200 系列高效液相色谱仪(VWD紫外检测器),美国安捷伦公司。
1.2 试药及试剂 诃子酸(批号wkq20032707,含量≥98%;购于四川维克奇生物科技有限公司);诃黎勒酸(批号wkq20042206,含量≥98%;购于四川维克奇生物科技有限公司);1,2,3,4,6-O-五没食子酰葡萄糖(批号14937-32-7,含量≥98%;购于成都普法瑞科技有限公司);没食子酸对照品(批号wkq16081904,含量≥98%;购于四川维克奇生物科技有限公司);柯里拉京对照品(批号wkq20032604,含量≥98%;购于四川维克奇生物科技有限公司);鞣花酸对照品(批号MO104AS,含量≥98%;购于美伦生物);纯净水(杭州娃哈哈集团有限公司),其余试剂均为分析纯;10批三果汤散由香格里拉藏医院制备,详细情况见表1。
表1 不同批次的三果汤散
2 方法与结果
2.1 溶液的制备
2.1.1 混合对照品溶液的制备 分别精密称取对照品GA、Corilagin、EA、CHI、CHA、PGG置于容量瓶中,用70%的甲醇溶解并定容至刻度,配置成浓度为0.43、0.32、0.23、0.58、0.23、0.58 mg/mL 的标准品溶液;取以上标准品溶液超声混匀,过 0.45 μm微孔滤膜,制备混合对照品溶液。
2.1.2 样品溶液制备 分别取10.0 g不同批次的三果汤散,加入150 mL水,浸泡30 min,于电热套上回流加热20、30、40、50 min,过滤,至旋转蒸发仪上旋干,温度为75 ℃,加入70%的甲醇 30 mL 溶解样品,至于50 mL离心管中,超声 20 min,离心3000 r,离心5 min,取上清液过0.45 μ微孔滤膜,即得40个样品溶液。
2.1.3 色谱条件 采用Agilent ZORBAX SB-C18色谱柱(4.6 mm×250 mm,5 μm),流动相为磷酸水溶液(A)-甲醇(B),梯度洗脱,检测波长273 nm,柱温25 ℃,流速1.0 mL/min。梯度见表2。

表2 洗脱程序
2.2 方法学考察 以样品S1为测试样品,按“3.1.2”煎煮30 min,进行精密度、稳定性、重复性实验,考察该分析方法的可靠性。
2.2.1 精密度实验 取S1样品,按“3.1.2”煎煮30 min处理样品,按“3.1.3”色谱条件连续进样6次,以4号峰(没食子酸)为参照峰,计算各色谱峰的相对保留时间和相对峰面积,结果14个共有峰的相对保留时间RSD小于0.2%,相对峰面积小于0.7%,表明仪器精密度良好。
2.2.2 重复性实验 取S1样品,按“3.1.2”煎煮30 min,制备样品6份,按“3.1.3”色谱条件分别进样,以4号峰(没食子酸)为参照峰,计算各色谱峰的相对保留时间和相对峰面积,结果14个共有峰的相对保留时间RSD小于0.21%,相对峰面积RSD小于0.55%,表明样品重复性较好。
2.2.3 稳定性实验 取S1样品,按“3.1.2”煎煮30 min制备样品,分别在0、2、4、6、8、10、12 h进样,以4号峰(没食子酸)为参照峰,结果14个共有峰的相对保留时间RSD小于0.6%,相对峰面积RSD小于0.8%,表明稳定性较好。
2.3 不同煎煮时间下10个批次的三果汤散指纹图谱结果分析
2.3.1 相似度评价 对不同批次的三果汤散进行指纹图谱分析,将图谱以“AIA”的格式导入“中药色谱指纹图谱相似度评价系统2012A(国家药典委员会)”软件,进行数据分析。以N1为参照图谱,时间窗宽度为0.1 min采用多点校正进行全谱峰匹配。选择5 min以后、文献报道较多的主要成分以及分离度较好的峰共14个共有峰。10批三果汤散指纹图谱匹配,计算相似度。见表3。

表3 不同煎煮时间下10批三果汤散的相似度评价结果
各批次的药物相似度都在0.9以上,说明该方剂品质均一稳定,差异不大,其中煎煮20、30 min时都以S8的相似度最低,40、50 min时以S7的最低,S1较好。
不同煎煮时间的三果汤对照图谱及叠加图谱如图1所示,其中N1~N10为煎煮20 min,N11~N20为煎煮30 min,N21~N30为煎煮40 min,N31~N40为煎煮50 min;混合对照品如图2所示,采用对照品指认了6个共有峰,分别是没食子酸酸(4)、柯里拉京(8)、诃黎勒酸(10)、1,2,3,4,6-O-五没食子酰葡萄糖(12)、诃子酸(13)、鞣花酸(14)。

图1 煎煮20、30、40、50min共有峰指纹图谱
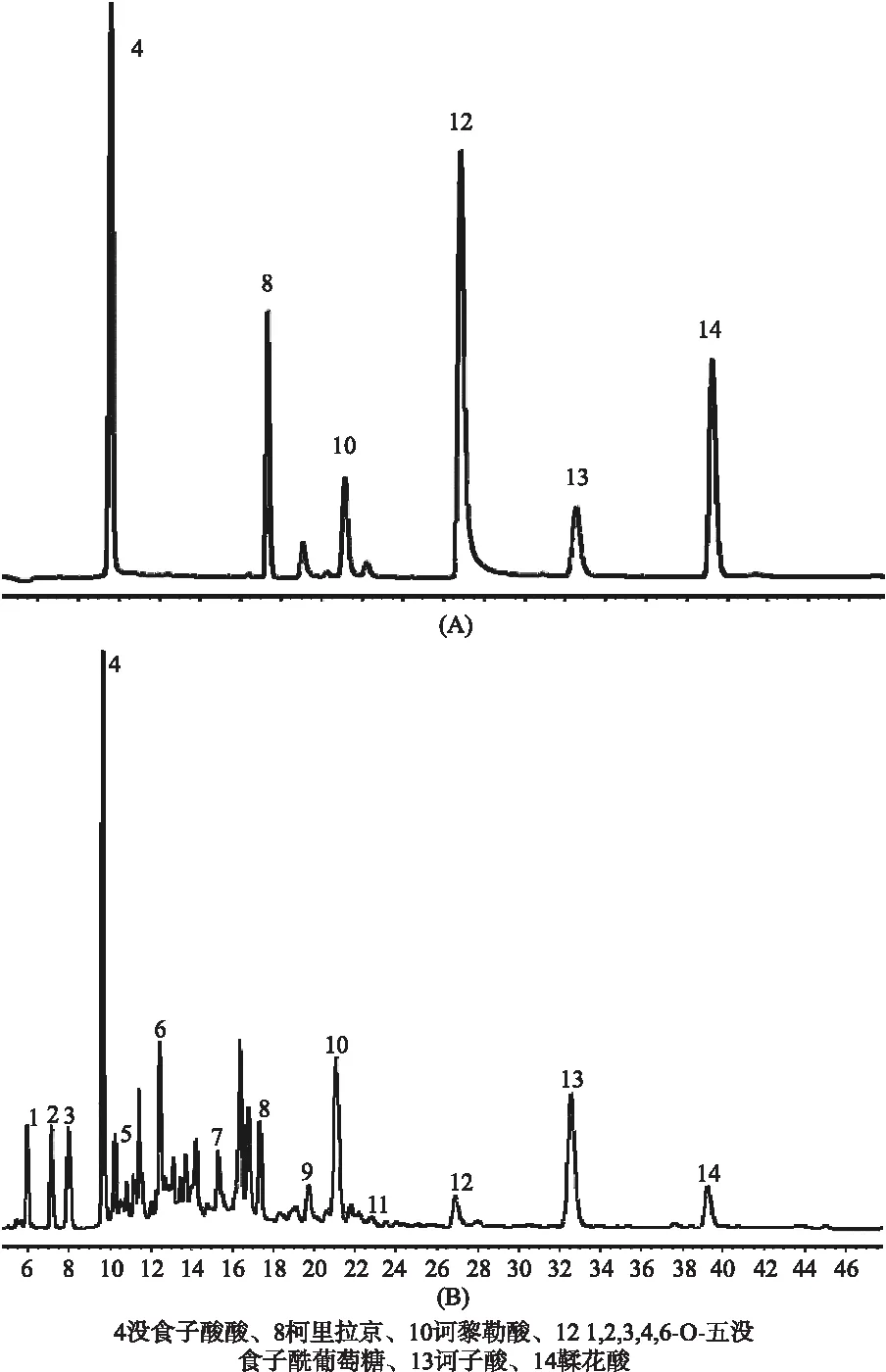
图2 A混合标准品色谱图,B样品谱图
2.3.2 聚类分析 将10批三果汤散样品的14个共有峰峰面积导入Origin2019b中的Cluster analysis中进行分析,CA显示可将样品分为四大类,N2、N6、N7、N8、N9、N10、N21、N22、N23、N28、N29、N32、N36、N37、N38、N39、N40共17个样品为第Ⅰ类,N13、N14、N15、N16、N17、N19、N20共7个样品为第Ⅱ类,N1、N3、N4、N5、N24、N25、N26、N27、N30、N31、N33、N34、N35共13个样品为第Ⅲ类,N11、N12、N18共3个样品为第Ⅳ类;如图3所示,其中从N1~N40为10批药材分别煎煮20~50 min,按序排。
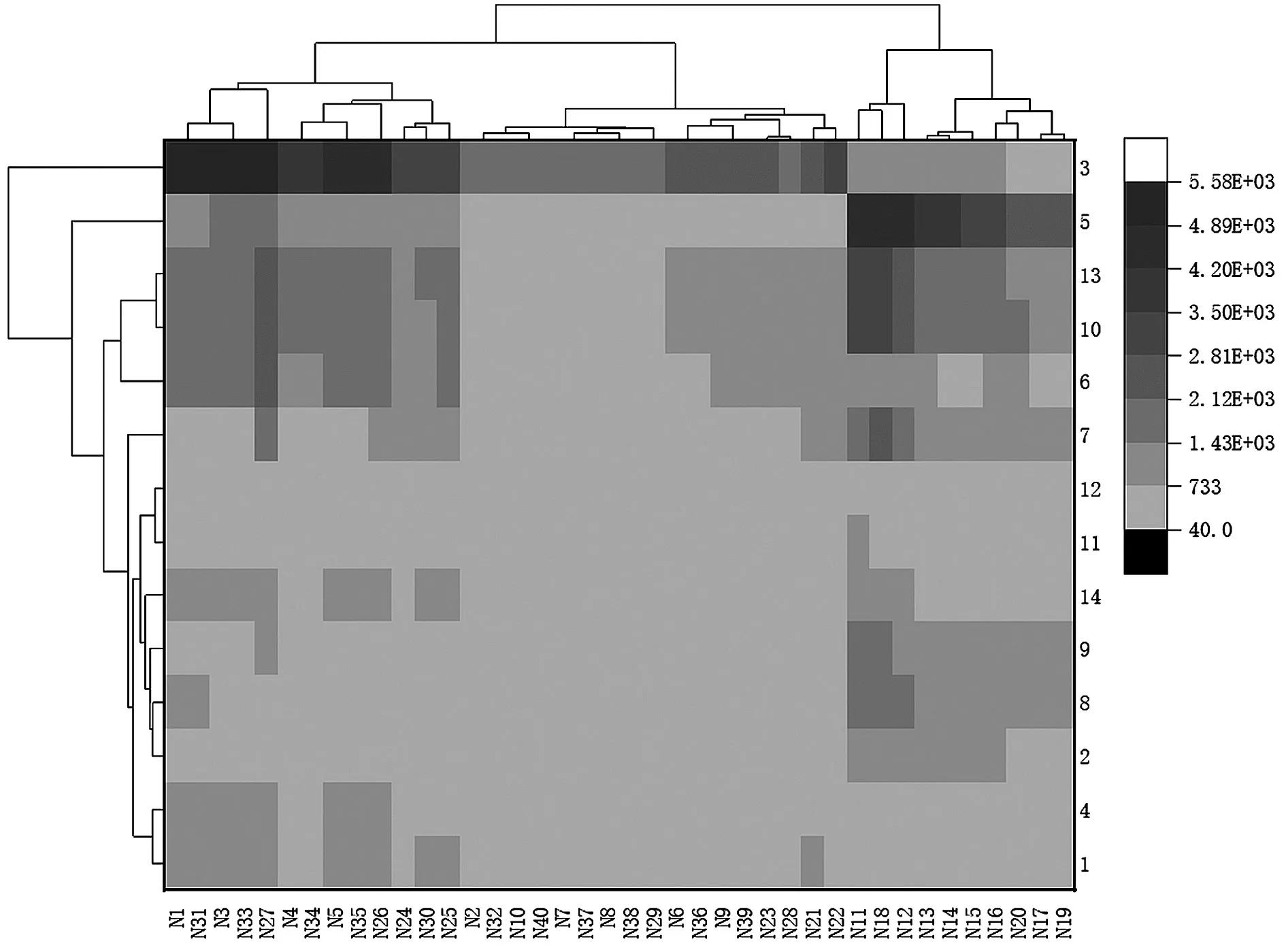
图3 不同煎煮时间下10批样品的14个峰共有峰聚类图
2.3.3 主成分分析 以14个共有峰为变量,将40个样品导入SIMCA-P 14.1 软件进行主成分(principal components analysis,PCA)分析。由图可知,得分矩阵图[R2X(cum)=0.992,Q2(cum)=0.967],表明该模型预测较好,可将40个样品区分为4类,该结果与CA一致。如图4所示。
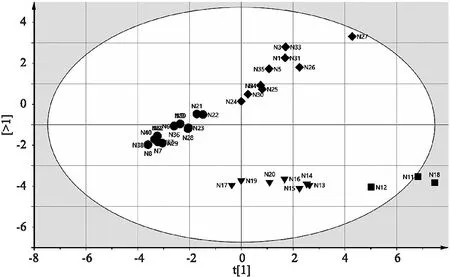
图4 不同煎煮时间下的10批样品的PCA得分图
2.3.4 正交偏最小二乘判别分析(OPLS-DA) 为了进一步检验样本是否有效或者样本间是否存在差异,找出对组分贡献较大的变量,将不同煎煮时间下10个批次样本共有的14个峰面积作为变量,导入SIMCA-P 14.1软件,采用OPLS-DA分析,观察样本聚集情况。其中R2X(cum)、R2Y(cum)和Q2(cum)分别是0.991、0.358、0.185,表明模型具有较好的预测性和稳定性,且其分类结果与PCA分析结果基本一致。

图5 不同煎煮时间下10批样品的OPLS-DA 得分图
载荷图(LoadingBi plot,如图6所示),峰1、3、4、6、12、14在第三类样品(N1、N3、N4、N5、N24、N25、N26、N27、N30、N31、N33、N34、N35)等样品周围,因此这些峰在第三类样品中贡献较大;剩下的峰8个峰与第四类样品(N11、N12、N18)样品最接近,因此剩下的8个峰对第四类样品聚类贡献较大,而第一类及第二类样品周围无共有峰分布,说明这两组样品中各组分的含量差异较小。

图6 不同煎煮时间下10批样品的OPLS-DA载荷图
在OPLS-DA模型中,为了筛选出样品中差异性较大的成分,使用变量权重值(Variableimportanceinprojection,VIP)评价方法,VIP>1.00的变量对组间分类贡献较大,具有统计学意义[17]。VIP>1.00的峰有5个:3号峰、5号峰、7号峰、10号峰(CHA)、13号峰(CHI)的VIP值分别为1.65、1.57、1.15、1.13、1.09,说明这几个峰对组间分类贡献较大。如图7所示。

图7 OPLS-DA 的变量权重图
3 讨论
3.1 色谱条件的选择 本实验先后考察了乙腈与磷酸水溶液、乙腈醋酸水溶液、甲醇甲酸水溶液的比例,最后选择了甲醇和0.1%的磷酸水溶液梯度洗脱可使标准品和样品达到较好的分离效果,酚酸鞣质类化合物在在273 nm时有较强的紫外吸收,因此选择273 nm作为指纹图谱的检测波长。
3.2 模式识别的分析 本实验建立不同煎煮时间对香格里拉藏医院的10批三果汤散的指纹图谱进行分析,显示样品都具有较高的相似性,在0.985~1之间。选择分离度较高,峰面积较大的色谱峰共14个,经对照品指认共有6个成分被分辨出来。不同煎煮时间的10批药材从直观结果分析显示,其样品含量较单一,药材质量无明显差异。CA与PCA分析结果显示可将不同煎煮时间下的10批三果汤散聚为4类,其分类情况存在一定的差异。并且通过OPLS-DA模型的载荷图、VIP值分析,判断14个峰对于样品分类的具体作用,从而判断出贡献最大的峰。
3.3 对不同煎煮时间下10批三果汤中6个化合物的相对峰面积变化的分析 由聚类分析发现,40个样本中的6个化合物可聚为两大类,说明GA的含量与其他5个成分差异较大。如图8所示。

图8 6个化学成分峰面积聚类分析图
以4号峰(没食子酸为对照),计算6个化合物的相对峰面积,并绘制曲线观察可知,在煎煮20、30、40、50min时间内,6个化合物的相对峰面积变化如图9所示:S1~S10中的Corilagin、EA、PGG的趋势基本一致,相对峰面积的大小也较稳定,CHA在S1~S6、S8、S10中先增后降,以30min达到最大值,其中又以S1和S8中最高,且CHI与CHA在S8中趋势一致,相对峰面积大小相当;CHI在S1~S7、S9~S10中都呈下降趋势,以20min达到最大值,其中又以S1、S9中最高。
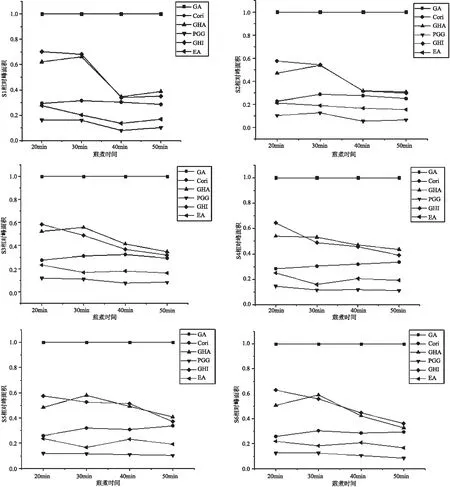

图9 不同煎煮时间下10批三果汤散中的6个化合物峰面积变化图
通过查阅文献分析可知,三果汤散的组成方药余甘子、诃子、毛诃子中都含有大量的酚酸鞣质化合物,尤其是如GA、CHI、CHA、PGG、1,3,6-三-O-没食子酰基葡萄糖等可水解鞣质[18]。水解鞣质在煎煮过程中,加热的时间可能会使某些化合物分解成GA。本实验将40个样品中的6个已知的化合物峰面积进行CA分析发现,GA与其他5个化合物存在较大差异,在所有样品中的聚集都是最明显的,因此随煎煮时间的增加,其他化合物可能会分解合成GA。峰面积折线图比显示10批三果汤散中以GA为参照,其他峰的峰面积比都小于1,差异变化较明显的峰是10号(CHA)与峰13号(CHI),除CHA外,其他5个化学成分都在20 min最高,综上可考虑选择20 min或30 min的煎煮时间为佳,但是不同煎煮时间下各成分的含量变化的相关性还需进一步研究。
目前三果汤指纹图谱全方位的质控研究报道较少,本实验建立不同煎煮时间10批三果汤散指纹图谱并结合化学模式分析的方法,操作便捷、方法稳定,可为三果汤散的质量控制及临床使用提供参考,但其临床应用还需后期药效学实验进一步研究。
