吡啶基有机碱催化O-羧基酐的活性开环聚合
2021-10-15李悦生
李 琛,李悦生
(天津大学材料科学与工程学院,天津300350)
脂肪族聚酯是一种可降解的环境友好材料,多数产品在12个月内即可完全分解,只释放出水和二氧化碳,对环境负担小[1~3].与此相反,聚乙烯、聚丙烯、聚氯乙烯和聚苯乙烯等传统塑料完全降解需要100年以上,大量堆积造成环境污染[4].脂肪族聚酯材料机械性能良好、生物相容性优异,在医用药载、产品包装和微电子等领域得到了广泛应用,是一种可部分替代传统塑料的新型高分子材料[1,3~5].环酯单体的开环聚合被广泛用于制备脂肪族聚酯材料[6].目前的研究主要集中在丙交酯和己内酯等环酯单体,所得聚合物链结构单一,力学性能有待进一步提高[1,7~16].
常见的单体修饰法包括扩环或缩环[7~9]、引入杂原子[10~13]和调节侧基等[14~16].研究表明,环酯单体的侧基修饰可有效调控聚酯材料的密度、黏度、结晶行为、热性能和力学强度等[15,17,18].1,3-二氧杂环乙烷-2,4-二酮,即O-羧基酐(OCA)是结构丰富的α-羟基酸衍生物,开环聚合制备的产物性能优异[15,19~23].与聚乳酸(PLA)相比,聚α-羟基苯乙酸的玻璃化转变温度(Tg)因苯环侧基的存在而大幅度提高,理论计算表明这种聚合物的力学性能可与聚苯乙烯媲美,综合性能优异[15].除乙交酯和丙交酯外,其它交酯单体因合成困难而难以得到实际应用.以α-羟基酸为原料时,侧基各异的OCA单体合成简便快捷,开环聚合后可获得各种结构和性能的聚α-羟基酸[24~26].
1951年,Davis[27]首次合成了OCA,但当时未被用于开环聚合.2006年,Bourissou等[28]使用4-二甲氨基吡啶(DMAP)催化Me-OCA的可控开环聚合.2014年,Guo等[29]报道了N-杂环卡宾催化OCA的可控开环聚合.2013年,Cheng等[30]用金属配合物催化OCA的可控开环聚合.近年来,OCA的开环聚合取得了重要进展,但仍难以有效抑制手性α-H的外消旋化,致使聚合物的立构规整度下降,力学性能变差[31,32].因此,探索可抑制酯交换和手性α-H外消旋化的新型高效催化剂具有重要意义[32~35].2014年,Lowe等[25]利用吡啶和羟基酸的复合物作为催化剂,获得了高立构规整度的聚α-羟基苯乙酸.2019年,Tao等[32]利用自制的硫脲基吡啶催化剂合成出全同立构规整度(Pm)高达90%的聚合物,解决了有机催化剂难以抑制手性α-H外消旋化的难题.本课题组致力于发展结构简单的廉价开环聚合催化剂,包括有机碱和Lewis pairs等[33].本文深入研究了环氧烷烃溶剂中吡啶基催化剂结构对OCA开环聚合的影响,通过结构优化获得一种高效廉价的有机催化剂,实现了OCA的活性开环聚合.
1 实验部分
1.1 试剂与仪器
膦腈碱t-BuP1购于百灵威试剂公司;4-甲氧基吡啶、4-甲基吡啶和DMAP购于美国Sigma-Aldrich试剂公司;吡啶、4-乙基吡啶、1,3-二异丙基硫脲、1,3-二苯基硫脲和三乙基硼购于英国Alfa试剂公司,使用前经过减压蒸馏纯化;L-扁桃酸、乳酸锂、L-(-)-3-苯乳酸、三光气、正丁基缩水甘油醚和超干四氢呋喃(THF)购于瑞士Adamas-Beta试剂公司;二氯甲烷、甲苯、正戊烷及乙醚等无水无氧溶剂由德国MBraun SPS溶剂纯化处理系统脱水精制;参照改进的文献方法合成苯基O-羧基酐(Ph-OCA)、甲基O-羧基酐(Me-OCA)和苄基O-羧基酐(Ph-OCA)[32,34];引发剂对苯二甲醇(BDM)购自Klamar试剂公司,使用前于80℃真空干燥6 h.
Bruker-400型核磁共振波谱仪(NMR),美国Bruker公司,测试温度25℃,以CDCl3为溶剂,四甲基硅烷(TMS)为内标;PL GPC-50型凝胶渗透色谱仪(GPC),上海沃特世科技有限公司,柱温40℃,流动相为色谱纯的四氢呋喃,流速为1.0 mL/min,以聚苯乙烯为标样校正.
1.2 OCA单体的制备
通过3种具有不同取代基的左旋α-羟基酸[L-(-)-3-苯乳酸、L-乳酸和L-扁桃酸]的缩合反应制备左旋Bnz-OCA、左旋Me-OCA和左旋Ph-OCA.在氮气气氛下,向盛有10 mmol左旋α-羟基酸和160 mg活性炭(120℃高温烘箱中活化2 h)的支口圆底烧瓶中加入500 mL超干四氢呋喃(超干四氢呋喃需大大过量,保证体系为假高稀溶液状态,否则会有黏浊液体低聚物产生,后期难以除去),然后将19.8 g(0.07 mol)三光气加入该混合物中,室温搅拌反应26 h;用砂芯漏斗过滤,收集有机溶液,并旋转蒸发,得到黄色固体粗产物;将黄色固体粗产物用1.0 L正戊烷反沉3次,敞口搅拌1 h,用砂芯漏斗过滤,收集固体产物;将固体产物溶解于四氢呋喃/乙醚混合溶剂中,放入冰箱中重结晶,重复4次操作后得到纯净的OCA白色固体,收率60%~85%.
左旋Bnz-OCA的1H NMR(400 MHz,CDCl3),δ:7.21~7.40(m,5H),5.31(t,1H),3.38(m,1H),3.27(dd,1H);13C NMR(100 MHz,CDCl3),δ:166.48,147.98,131.66,129.77,129.26,128.50,80.03,36.47.
左旋Me-OCA的1H NMR(400 MHz,CDCl3),δ:5.12(q,1H),1.71(d,3H);13C NMR(100 MHz,CDCl3),δ:167.76,148.20,76.29,16.60.
左旋Ph-OCA的1H NMR(400 MHz,CDCl3),δ:7.42~7.53(m,5H),6.02(s,1H);13C NMR(100 MHz,CDCl3),δ:165.46,148.15,130.92,129.67,129.34,126.26,80.55.
2 结果与讨论
2.1 催化剂结构选择
首先选择四氢呋喃和甲苯作为反应溶剂,在60℃下尝试用磷腈碱t-BuP1作为催化剂.使用活性更高的BDM引发Bnz-OCA开环聚合,发现聚合活性较低.反应6 min,THF组的单体转化率为13%,催化剂转换频率(TOF)为68.5 h-1,分子量分布较宽(Đ=1.64);甲苯组的单体转化率为15%,TOF为78.7 h-1.为提高OCA的聚合活性,尝试用强极性的正丁基缩水甘油醚(NBGE)作为反应溶剂.前期工作中,我们曾用NBGE作为Lewis pair催化丙交酯活性开环聚合的反应介质,发现效果明显优于THF等溶剂[35].在6 min内,Bnz-OCA的转化率高达74%,TOF为375.6 h-1,分子量分布指数为Đ=1.44.改用NBGE溶剂后,Bnz-OCA的反应活性提高了5倍,但聚合物的分子量分布仍然较宽.以上结果表明,使用磷腈碱t-BuP1催化OCA开环聚合可控性差.
Lewis pairs,包括布朗斯特酸/Lewis碱对,已被证明可抑制丙交酯和己内酯等环酯单体开环聚合的酯交换副反应[32].因此,本文用各种Lewis pairs作为催化剂,希望能改善OCA开环聚合的可控性(Scheme 1).在60℃的NBGE溶剂中,用BEt3/t-BuP1、1,3-二苯基硫脲TU1/t-BuP1、1,3-二异丙基硫脲TU2/t-BuP1催化Bnz-OCA的开环聚合,代表性实验结果列于表1.实验数据(Entries 2~4)表明,Lewis pairs催化聚合产物的分子量分布有所变窄,但效果不明显,说明这些酸碱对催化剂并不能完全抑制OCA开环聚合的酯交换副反应.
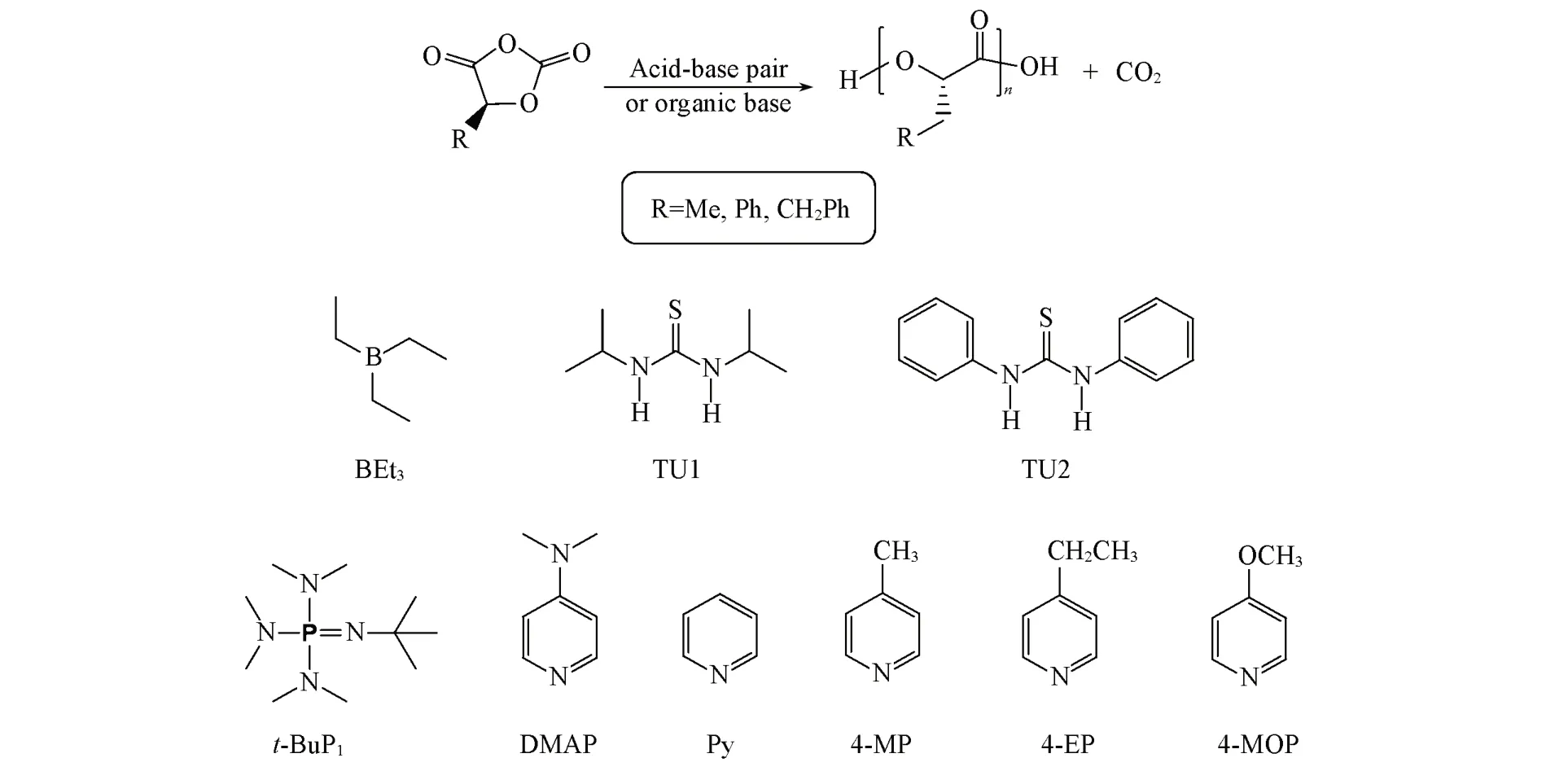
Scheme 1 Ring⁃opening polymerization of various OCAs and the catalysts used
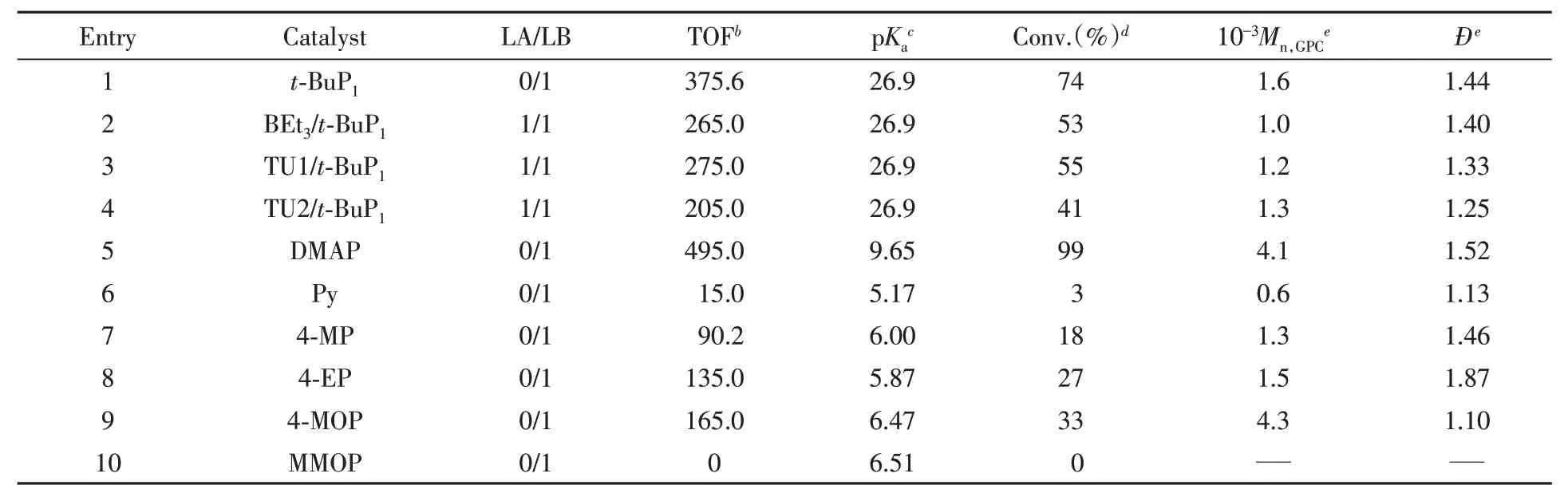
Table 1 Ring-opening polymerization of Bnz-OCA by various catalystsa
对比表1中Entry 5和Entry 1的实验数据可以发现,在相同条件下DMAP的催化活性明显高于t-BuP1,所以本文的后续工作聚焦于吡啶基Lewis碱.于60℃用DMAP催化Bnz-OCA的开环聚合,6 min单体转化接近完全,TOF高达495.0 h-1,但酯交换等反应严重,分子量分布指数大于1.5,GPC曲线呈现不规则的三峰分布(图1).DMAP不能有效抑制酯交换副反应的原因可能是其碱性仍然较强(共轭酸的pKa=9.65).为了验证这一推测,本文用低碱性的吡啶(共轭酸的pKa=5.17)催化Bnz-OCA的开环聚合.虽然在6 min内Bnz-OCA的转化率只有3%,TOF为15.0 h-1,仅为DMAP的1/33,但所得聚合物的分子量分布很窄,Đ=1.13(Entry 6,表1),表明酯交换等副反应得到了有效抑制.吡啶催化活性不高的可能原因是碱性过低,活化OCA的羰基速率较慢,使OCA脱CO2开环聚合比较困难.
上述结果表明,环氧化物溶剂中OCA开环聚合的理想Lewis碱催化剂的碱性应介于吡啶和DMAP之间,这样的Lewis碱既可催化OCA的快速活性聚合,又能有效抑制酯交换副反应,生成高分子量、窄分子量分布的聚α-羟基酸.为了调控吡啶衍生物的碱性,向吡啶环的4号位引入供电子效应低于—NMe2的取代基,包括甲基(Me)、乙基(Et)和甲氧基(OMe).
在60℃下,4-甲基吡啶催化Bnz-OCA开环聚合的活性明显高于吡啶,6 min内单体转化率为18%,TOF为105.0 h-1,但分子量分布没有明显变化(Đ=1.46),类似于DMAP的情况.乙基的供电子强度高于甲基,4-乙基吡啶的碱性更高,催化Bnz-OCA开环聚合速率变快,6 min内单体转化率达到27%.严重的酯交换副反应使聚合物分子量分布较宽(Đ=1.87).以上结果表明,4-甲基吡啶和4-乙基吡啶并不是催化OCA开环聚合的合适催化剂.
烷基的供电子效应来源于供电子诱导和σ-π超共轭效应,远程传递效果较差;而烷氧基的p-π共轭效应供电能力强,远程传递效果好,有利于加速催化反应的电子转移过程.因此,本文尝试用4-甲氧基吡啶(4-MOP)催化Bnz-OCA的开环聚合.结果表明,4-MOP不仅具有高催化活性,而且还可有效抑制酯交换副反应,催化聚合体系具有明显的活性聚合特征.Bnz-OCA在6 min内转化率可达到33%,TOF为165 h-1(Entry 9,表1).由图1可见,聚合物的GPC淋出曲线呈现完美的单峰分布,分子量分布指数Đ=1.10,测得聚合物分子量为4300,接近理论预测值.
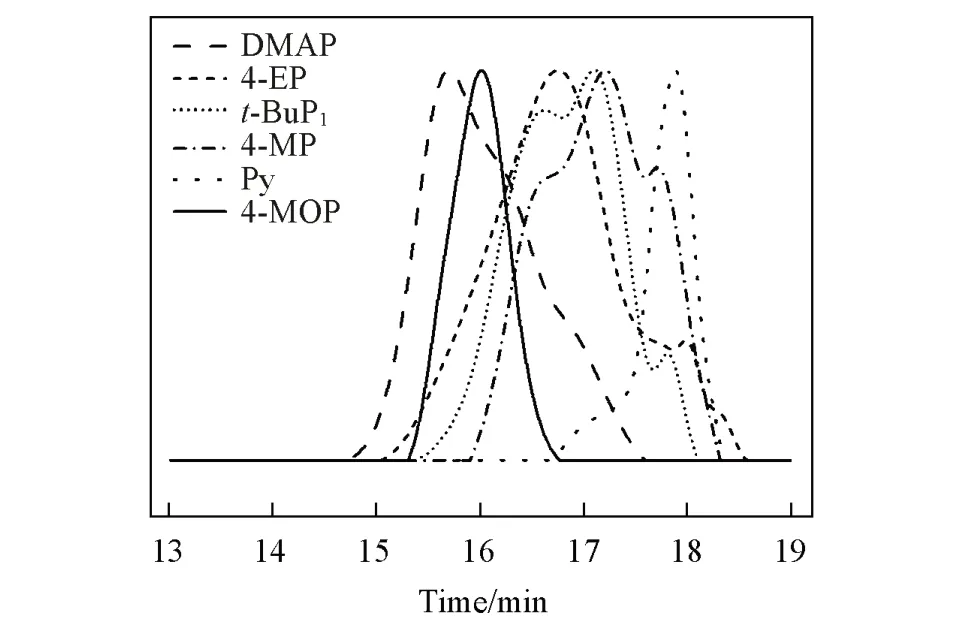
Fig.1 GPC trace of polyhydroxyphenylpropionic acid by different catalysts
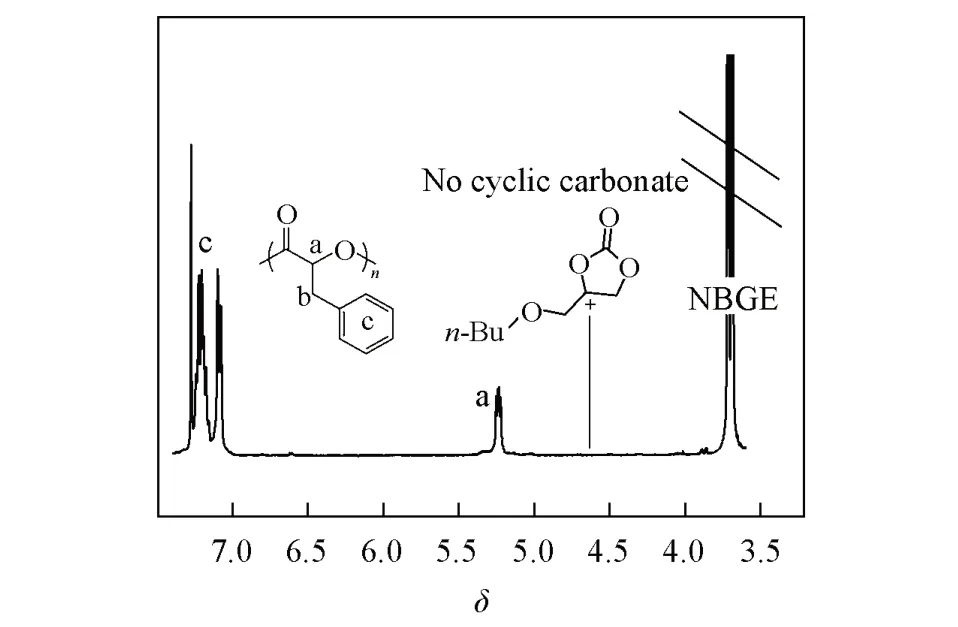
Fig.2 1H NMR spectrum of the reaction mixture of OCA polymerization by 4⁃MOP
不同于丙交酯等其它环酯单体,OCA的开环聚合伴随着小分子CO2的释放.大多数开环聚合催化剂也能促进CO2与环氧烷烃的偶联环化或交替共聚合反应,所以在环氧烷烃中进行OCA的开环聚合时可能伴随着环碳酸酯或脂肪族聚碳酸酯副产物的生成.图2给出4-MOP催化Bnz-OCA开环聚合溶液的1H NMR图谱.在δ4.6处未观测到特征质子共振信号,表明OCA开环聚合产生的CO2没有发生相应的后续副反应.在现有文献中,这一现象较少见.另外,4-MOP来源方便、价格低廉、安全无毒.以上结果说明,4-MOP是OCA开环聚合的理想催化剂,具有良好的应用前景.
吡啶类Lewis碱催化剂按催化活性依次降序排列如下:4-二甲氨基吡啶(DMAP)>4-甲氧基吡啶(4-MOP)>4-乙基吡啶(4-EP)>4-甲基吡啶(4-MP)>吡啶(Py).将TOF对催化剂共轭酸的pKa值作图,得到线性关系图(图3).可以发现,TOF与共轭酸的pKa值成正比,即随着碱强度增大,催化活性线性增大.拟合直线的决定系数R2=0.981,表现出很强的规律性.TOF对催化剂共轭酸的pKa值符合公式:TOF=100.96pKa−482.48.
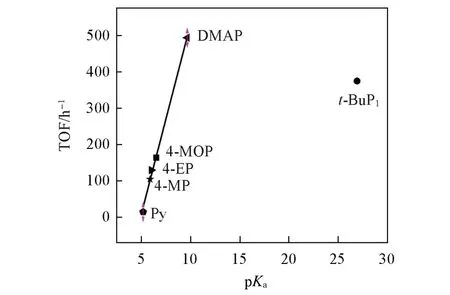
Fig.3 Relationship of TOF vs.pKa in ROP of Bnz⁃OCA by organic bases
2.2 聚α⁃羟基酸的立构规整性
天然来源的α-羟基酸是手性化合物,手性中心是α-碳原子.手性α-羟基酸与三光气的反应不涉及手性中心α-碳原子,因此OCA也是手性化合物.如果有机催化剂的碱性较强,则可与弱酸性的α-H反应,生成平面结构的酯烯醇负离子;酯烯醇负离子的两侧均可与有机碱的共轭酸反应,生成外消旋OCA(Scheme 2).即有机强Lewis碱可催化手性OCA的外消旋化反应.因此,用有机Lewis碱催化OCA的开环聚合有可能引发手性α-H的外消旋化反应,外消旋化程度取决于催化剂的碱性和反应条件.在优化反应条件下,使用弱碱性催化剂可完全抑制外消旋化反应,合成高等规度的聚α-羟基酸.
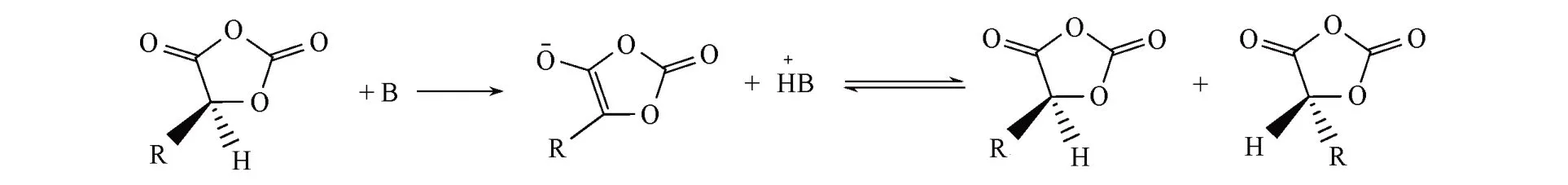
Scheme 2 OCA racemization by base catalysis
由图4可见,用强碱t-BuP1和DMAP催化Bnz-OCA的开环聚合,根据公式Immm=Pm(Pm+1)/2(Immm指全同mmm四元组积分面积与所有四元组的积分面积和的比值)[31]计算出聚合物全同立构规整度Pm分别为0.17和0.55,均没有得到高立构规整度的聚α-羟基酸,聚合物上的手性α-H的特征共振信号峰裂分不规则,外消旋化副反应严重.用低碱性的吡啶催化Bnz-OCA的开环聚合,Pm=0.81,所得聚合物上的手性α-H的特征共振信号峰裂分为稍显不对称的四重峰,说明该聚合过程中没有明显的外消旋化副反应发生.用4-MOP催化Bnz-OCA的开环聚合时,所得聚合物上的手性α-H的特征共振信号峰裂分为完全对称的四重峰,mmm全同四元组的积分面积占比93%,全同立构规整度Pm高达0.90,说明几乎完全消除了α-H的外消旋化副反应.
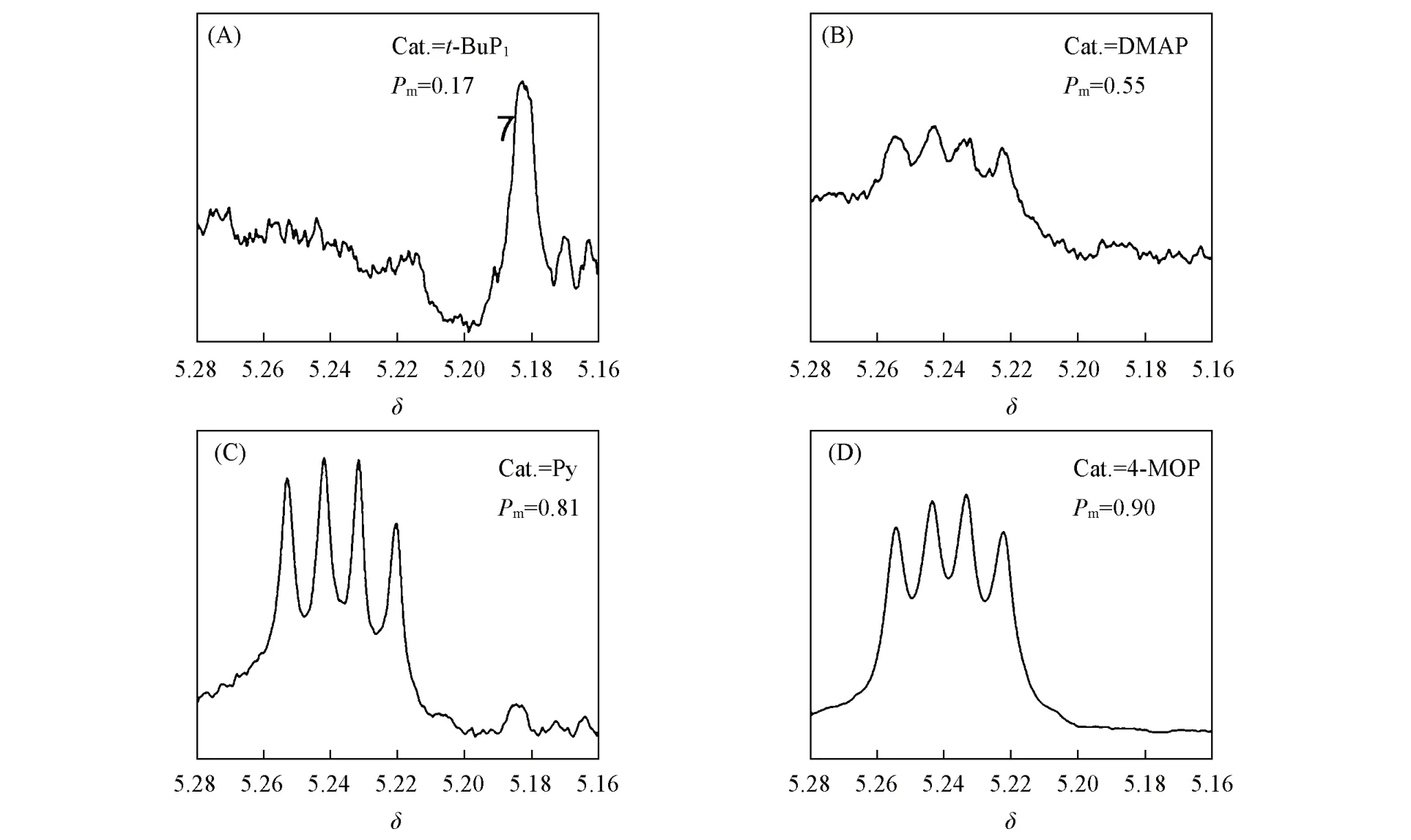
Fig.4 α⁃H resonance signal peaks of polymers by different catalysts
2.3 不同OCA的开环聚合
由于4-MOP表现出优异的催化性能,进一步在60℃的NBGE中用其催化Me-OCA,Bnz-OCA和Ph-OCA开环聚合,代表性实验结果列于表2.Bnz-OCA在20 min内即可转化完全,聚合物的分子量分布指数仅为1.08.Me-OCA的聚合活性与Bnz-OCA非常接近,生成分子量分布较窄的聚乳酸(Đ=1.16).在相同条件下,Ph-OCA的开环聚合速率较慢,需要2 h才能转化完全.不同于Bnz-OCA,Ph-OCA分子中苯环直接与α-碳原子相连,使羰基周围空间位阻较大,所以Ph-OCA的聚合活性比较低.实验数据(Entries 3~5,表2)表明,由于催化聚合体系具有活性聚合特征,增大单体和引发剂的投料比并延长反应时间可以高效合成高分子量的聚α-羟基酸,同时保持很窄的分子量分布.
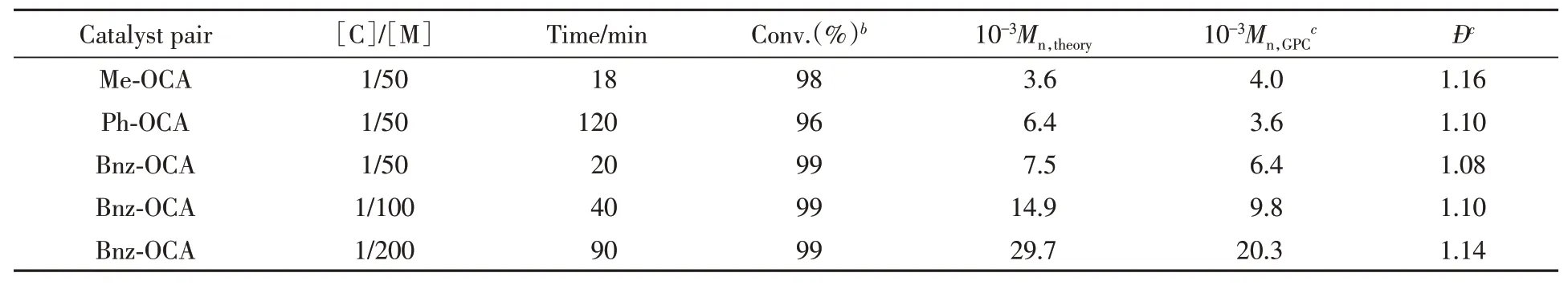
Table 2 Ring-opening polymerization of OCAs by 4-MOPa
2.4 聚合反应动力学
为了进一步理解4-MOP催化OCA开环聚合的反应过程,研究了环氧化物溶剂NBGE中OCA开环聚合的动力学.以Bnz-OCA转化率(C)对反应时间作图得到一条直线,动力学方程为dC/dt=0.05,决定系数R2=0.98,即Bnz-OCA的转化率与反应时间成正比;如果以ln{[OCA]0/[OCAt]}对反应时间作图,数据点明显不落在同一条直线上(图5),表明4-MOP催化OCA的开环聚合遵循零级动力学,即开环聚合速率与单体浓度无关.在四氢呋喃或甲苯中用4-MOP催化Bnz-OCA的开环聚合时,聚合速率与单体浓度关系遵循一级动力学.这一实验结果表明,环氧烷烃可能参与了OCA的活化开环过程,但反应机理尚有待进一步研究.
为了揭示环氧烷烃结构对开环聚合的影响,除NBGE外,还研究了OCA在烯丙基缩水甘油醚(AGE)和氧化苯乙烯(SO)中的开环聚合反应动力学.由图6可见,OCA在不同环氧化物溶剂中的开环聚合反应均遵循零级动力学,并且聚合速率几乎不受环氧化物溶剂种类的影响,说明溶液分子中的环氧基团对OCA开环聚合起到了促进作用.
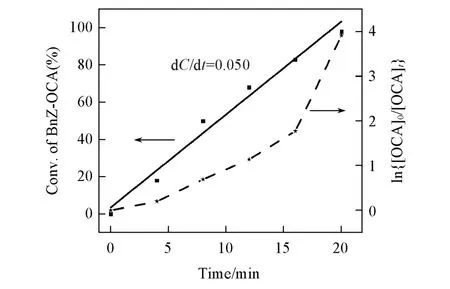
Fig.5 Plots of Bnz⁃OCA conversion vs.reaction time in NBGE obtained by 1H NMR
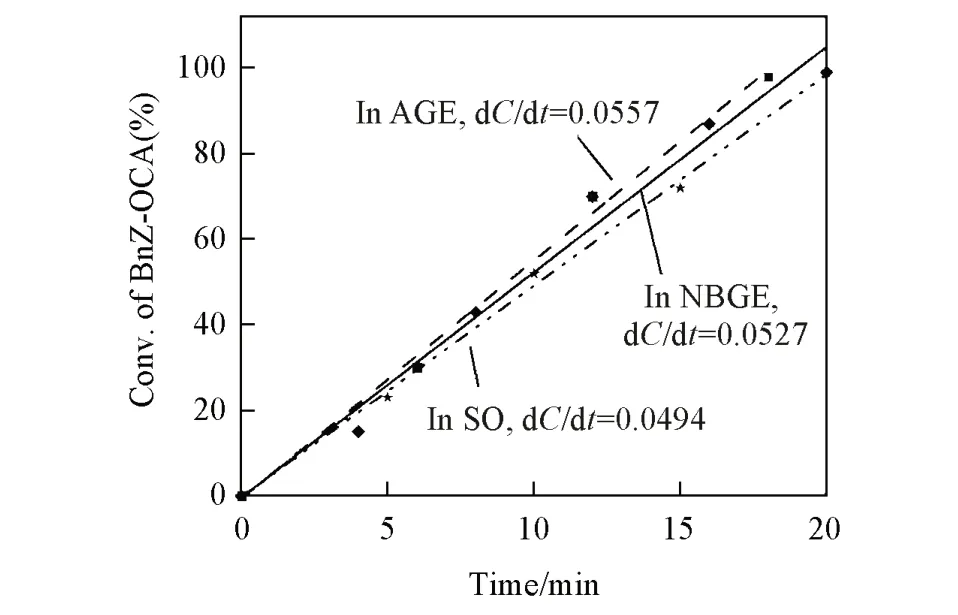
Fig.6 Plots of Bnz⁃OCA conversion vs.reaction time in different solvents obtained by 1H NMR
3 结 论
膦腈碱t-BuP1和DMAP等有机强碱在环氧烷烃溶剂中可快速催化OCA的开环聚合,但酯交换副反应和手性α-H的外消旋化反应严重,聚合反应的可控性差.有机强碱催化聚合体系组合布朗斯特酸或Lewis酸仍不能有效抑制酯交换和α-H外消旋化反应.在DMAP基础上降低吡啶衍生物的碱性,可抑制酯交换和手性α-H的外消旋化反应,提高开环聚合的可控性和聚α-羟基酸的立构规整度.吡啶衍生物的TOF对碱强度(共轭酸的pKa值)呈线性关系,催化剂碱性越低聚合速率越小,但可控性增强.碱性适中的4-MOP是OCA开环聚合的理想催化剂,在60℃的环氧化物溶液中,TOF可达到165 h-1,聚合物的分子量分布指数可控制在1.10左右,具有明显的活性聚合特征,聚合物的等规度可控制在90%以上.虽然环氧化物是高活性物质,但在有机碱的催化聚合过程中并不与OCA开环释放的CO2发生反应.OCA在甲苯和四氢呋喃等溶液中进行开环聚合呈一级动力学,聚合速率与单体浓度成正比;然而,OCA在环氧烷烃溶液中的开环聚合遵循零级动力学,即聚合反应速率与单体浓度无关.可能原因是环氧烷烃参与了OCA的活化开环过程,具体反应机理有待进一步研究.
