泪腺IgG4相关性眼病19例临床病理分析
2021-10-12杨菁茹任雨洁李元朋夏益敏王卉芳蒋婉婷
刘 辉,杨菁茹,任雨洁,李元朋,夏益敏,王卉芳,蒋婉婷
IgG4相关性疾病(IgG4-related disease, IgG4-RD)是近年才逐渐被认识的一类自身免疫性系统性疾病[1-3],病变累及眼部时称为IgG4相关性眼病(IgG4-related ophthalmic disease, IgG4-ROD)。IgG4-ROD常累及泪腺、眼外肌、眼睑及三叉神经等[4],泪腺IgG4-ROD常表现为双眼泪腺无痛性肿大,病理学上常需要与泪腺炎性假瘤、Mikuliczs病和淋巴瘤等相鉴别。本文回顾性分析19例泪腺IgG4-ROD,并收集泪腺炎性假瘤和Mikuliczs病作为对照,分析其临床表现、影像学特征及病理学改变并复习文献,以提高临床及病理医师对泪腺IgG4-ROD的认识水平。
1 材料与方法
1.1 材料收集陕西省西安市人民医院(西安市第四医院)2010年12月~2020年12月确诊的泪腺IgG4-ROD 19例(32只眼),另收集10例泪腺炎性假瘤和10例泪腺Mikuliczs病作为对照(22只眼)。切片均由两位经验丰富的病理医师重新阅片,并收集所有病例的临床资料进行分析。
1.2 方法组织均经10%中性福尔马林固定,石蜡包埋,4 μm厚连续切片,免疫组化染色采用EnVision法,所用IgG4和IgG一抗均购自福州迈新公司,具体操作步骤严格按试剂盒说明书进行。
1.3 病理学特征判断病理指标判读标准如下。淋巴滤泡形成程度判断:每HPF可见3个及以上淋巴滤泡为“”,每HPF可见2个淋巴滤泡为“”,每HPF可见1个淋巴滤泡为“+”,未见淋巴滤泡为“-”。间质纤维化程度判断:>1/2泪腺组织被纤维组织替代为“”,1/3~1/2泪腺组织被纤维组织替代为“”,<1/3泪腺组织被纤维组织替代为“+”,未见纤维组织为“-”。根据是否出现席纹状纤维化、嗜酸性粒细胞及闭塞性静脉炎相应判读为阳性或阴性。
1.4 诊断标准实验组病例必须满足:(1)影像学检查发现泪腺肿大、三叉神经增粗、眼外肌增粗或包块,或眼部组织多发的肥厚性病变;(2)组织病理学发现大量淋巴细胞和浆细胞浸润,有时伴有纤维化,通常会有一个生发中心,且满足IgG4/IgG细胞>40%且IgG4阳性浆细胞>50个/HPF。此外当血清IgG4≥1.35 g/L为“确定诊断”,无血清IgG4升高时仅满足条件(1)和(2)时为“很可能诊断”,本文将“确定诊断”和“很可能诊断”均纳入实验组。
1.5 统计学方法采用SPSS 22.0软件进行统计学分析,组织病理学特征等计数资料组间比较采用χ2检验;间质纤维化程度及淋巴滤泡数量属于等级资料,组间比较采用Wilcoxon秩和检验;IgG4阳性浆细胞数为计量资料,呈非正态分布以M(P25,P75)表示,组间比较采用Wilcoxon秩和检验,以P<0.05为差异有统计学意义。
2 结果
2.1 临床及影像学特征19例泪腺IgG4-ROD患者年龄28~83岁,平均57.4岁,男女比为12 ∶7,病变位于双眼13例,单眼6例,临床表现多为泪腺无痛性肿大,部分患者伴有眼球突出或运动受限,其中14例患者血清IgG4升高(3.33~21.7 g/L)。影像学多表现为双眼泪腺肿大,部分患者出现眼外肌肿大或眶下神经增粗。部分患者出现病情反复,其中最长病程反复发作持续14年,19例IgG4-ROD中14例获得随访,随访时间5个月~4年,6例复发。
2.2 病理特征9例泪腺IgG4-ROD大体多表现为境界清楚的结节,切面实性,质中,部分送检组织破碎。镜下表现为显著的淋巴细胞及浆细胞浸润,淋巴滤泡形成(图1),背景常伴有显著的纤维化,并可见席纹状纤维化(图2),闭塞性静脉炎未见,免疫组化可见大量IgG(图3)和IgG4阳性浆细胞(图4),且均满足IgG4/IgG细胞>40%及IgG4阳性浆细胞>50个/HPF。对照组(泪腺炎性假瘤和Mikuliczs病)病例中也可见显著的纤维化及大量淋巴细胞浸润,淋巴滤泡形成,但并未发现大量IgG4阳性浆细胞,仅表现为散在少量IgG4阳性浆细胞(图5、6)。
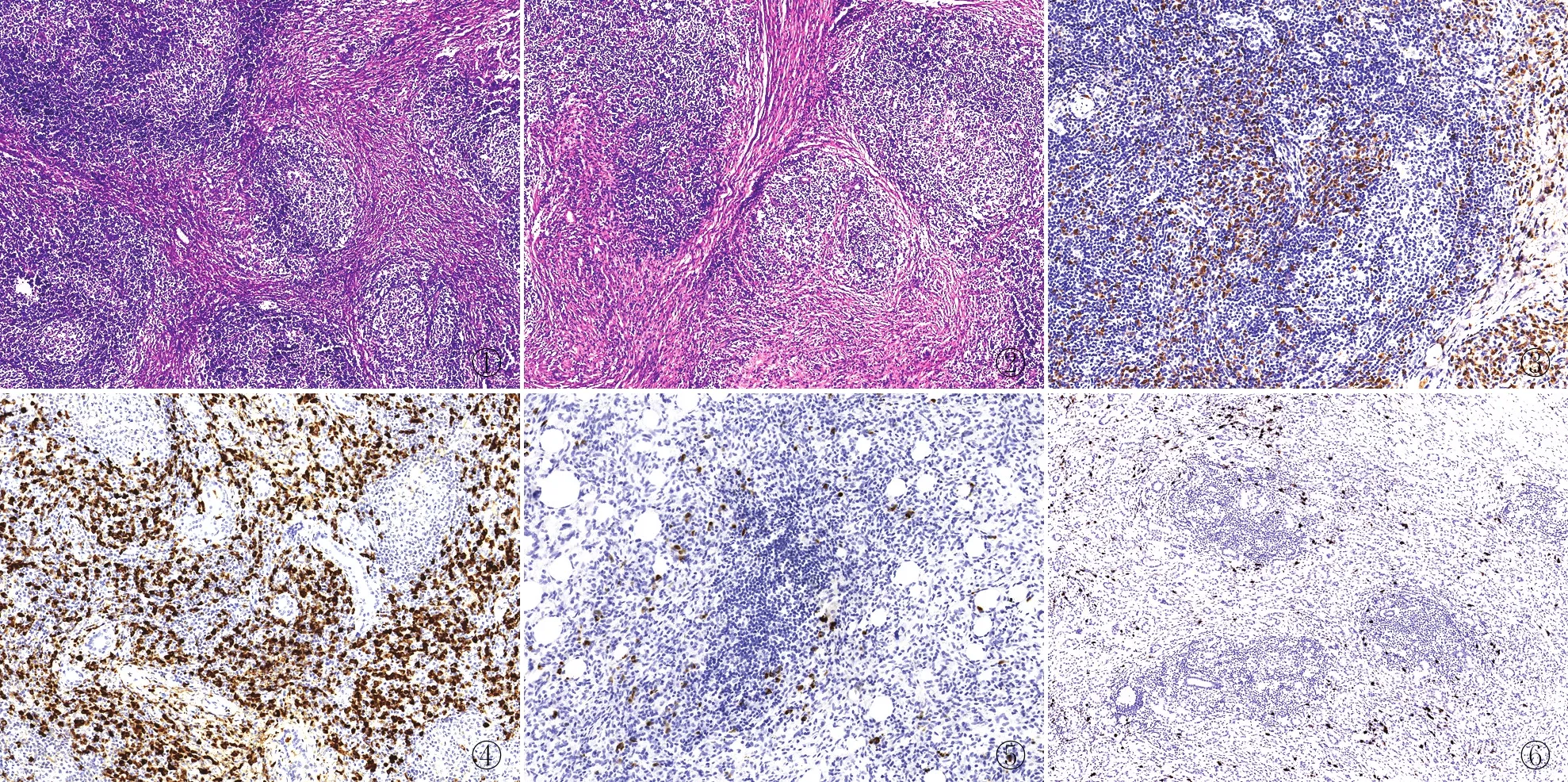
图1 泪腺IgG4-ROD镜下见显著的淋巴细胞及浆细胞浸润,淋巴滤泡形成 图2 泪腺IgG4-ROD镜下见背景伴有显著的纤维化,并出现席纹状纤维化 图3 泪腺IgG4-ROD病灶组织中出现IgG阳性浆细胞,EnVision法 图4 泪腺IgG4-ROD病灶组织中出现大量IgG4阳性浆细胞,EnVision法 图5 泪腺炎性假瘤病变组织中仅出现散在少量IgG4阳性浆细胞,EnVision法 图6 泪腺Mikuliczs病变组织中仅出现散在少量IgG4阳性浆细胞,EnVision法
2.3 泪腺IgG4-ROD组与非IgG4-ROD组临床病理特征比较泪腺IgG4-ROD组与非IgG4-ROD组相比,性别差异无显著性(P>0.05)。IgG4-ROD组病变双眼累及率更高,差异有显著性(P<0.05)。IgG4-ROD组淋巴滤泡形成更为明显,均为中到重度(~),与对照组相比差异有显著性(P<0.05)。IgG4-ROD组背景纤维化更为明显,中到重度纤维化病例更多见(~),差异有显著性(P<0.05)。IgG4-ROD组IgG4阳性浆细胞数量中位数为60个/HPF,对照组中位数仅为1个/HPF,差异有显著性(P<0.05,表1)。
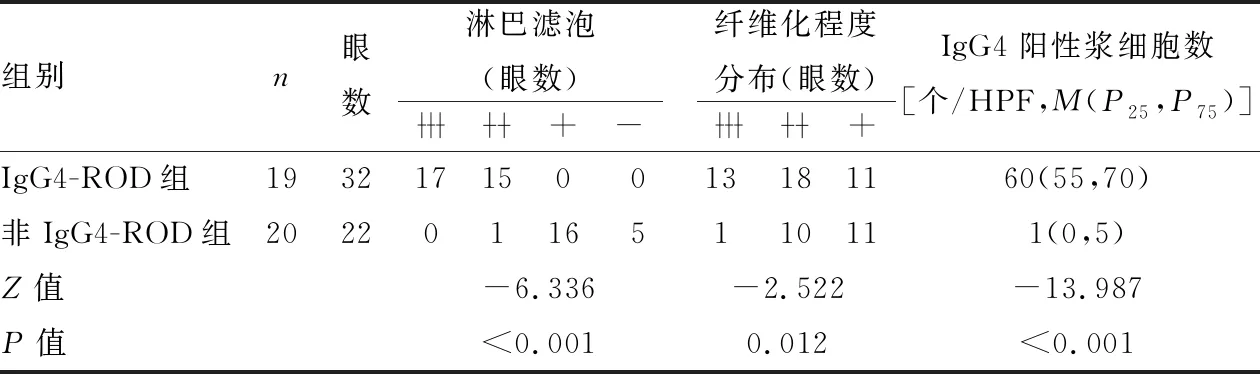
表1 泪腺IgG4-ROD组与非IgG4-ROD组病理特征比较
3 讨论
随着2015年Goto等[5]提出的IgG4-ROD诊断标准逐渐被采纳,人们对于该类疾病的认识也逐渐深入,部分曾经诊断的泪腺淋巴组织增生性疾病被重新认识分类。IgG4-ROD占系统性IgG4-RD的4%~34%[6],好发于中老年人,女性多见,常累及泪腺、眼外肌、眼睑及三叉神经等,也有罕见报道发生于巩膜和葡萄膜等眼内组织[6-9]。泪腺IgG4-ROD主要表现为双侧泪腺无痛性肿大,影像学改变主要为泪腺肿大、眶下神经增粗及眼外肌肿大。病理学表现为大量淋巴细胞和浆细胞浸润,伴或不伴有纤维化,常可见淋巴滤泡形成,免疫组化标记IgG4阳性浆细胞>50个/HPF且IgG4/IgG阳性细胞>40%。大部分IgG4-RD患者血清IgG4升高(>1.35 mg/L),但临床研究发现确实有部分病例存在典型IgG4-RD病理学和影像学改变,但血清IgG4并不升高[6]。因此,2015年Goto等[5]提出当血清IgG4水平并无升高时,满足其他2个诊断指标时将其归为“很可能诊断”[5],既往多数学者将符合“很可能诊断”标准的病例纳入IgG4-ROD组进行研究。本组19例IgG4-ROD中,包含5例血清IgG4并无升高的符合“很可能诊断”标准的患者。IgG4-ROD与IgG4-RD诊断标准的主要区别在于IgG4阳性浆细胞数量的界定,IgG4-RD诊断标准中要求IgG4阳性浆细胞>10个/HPF,而IgG4-ROD则要求IgG4阳性浆细胞>50个/HPF,其差异性是基于眼及附属器病变时常常发生成熟淋巴细胞增生性病变,且可出现部分浆细胞IgG4阳性,也有学者提出达到30个/HPF即可诊断IgG4-ROD,但目前普遍认为IgG4阳性浆细胞>50个/HPF诊断IgG4-ROD更为保险。IgG4-ROD与其他器官IgG4-ROD病理学特征区别还在于IgG4-ROD中闭塞性静脉炎并不常见[6],本组19例IgG4-ROD中均未发现典型的闭塞性静脉炎,但纤维化明显且淋巴滤泡数量显著增多。
泪腺常见的淋巴组织增生性病变包括泪腺炎性假瘤、Mikuliczs病、淋巴组织反应性增生及淋巴瘤等[10]。IgG4-ROD也属于淋巴组织增生性病变,这类疾病镜下均表现为成熟小淋巴细胞浸润伴或不伴有间质纤维化,相似的镜下表现且基于眼眶部位限制性,往往无法送检完整病变组织,特别是当送检标本为小块破碎组织时,往往造成鉴别诊断困难。回顾性研究发现以往曾经诊断为眼部炎性假瘤、Mikuliczs病和慢性非特异性泪腺炎的部分病例应归类为IgG4-ROD。本组19例IgG4-ROD中包含曾经被诊断例泪腺炎性假瘤4例和Mikuliczs病5例,还包含1例曾被诊断为泪腺结外边缘区黏膜相关淋巴组织淋巴瘤病例,因而泪腺IgG4-ROD与泪腺炎性假瘤和Mikuliczs病等常见淋巴组织增生性疾病的鉴别诊断尤为重要。
泪腺炎性假瘤属于特发性慢性非特异性泪腺炎,多为单侧发病,可广泛累及眼部软组织,部分病例病灶与周围组织界线不清,患者常表现为眼睑肿胀、疼痛及流泪等急性炎性症状,本实验收集的泪腺炎性假瘤患者大多表现为眼部疼痛及压痛,部分患者伴有泪腺肿大,病理学表现为弥漫性淋巴细胞及浆细胞浸润,可见成熟淋巴滤泡形成,可伴有间质纤维化和肌纤维母细胞增生,部分病例可见胶原纤维堆积。炎性假瘤作为一种排它性诊断,需除外其他疾病才能确诊,基于其临床表现、影像学和病理学特征均与IgG4-ROD类似,特别是当患者未出现眼部急性炎症症状时,主要依靠免疫组化染色进行鉴别,炎性假瘤病变组织未见或仅有少量IgG4阳性浆细胞。由于IgG4-ROD概念是近几年才被提出,且炎性假瘤病变组织中也可出现IgG4阳性浆细胞,回顾性研究发现曾经诊断为泪腺炎性假瘤的部分病例应归类为IgG4-ROD[11]。本组19例IgG4-ROD中4例曾经诊断为泪腺炎性假瘤,免疫组化检测发现存在大量IgG4阳性浆细胞,符合IgG4-ROD诊断标准被重新分类;对照组10例泪腺炎性假瘤,其中5例出现散在IgG4阳性,但并未达到IgG4-ROD诊断标准中要求的﹥50个/HPF,也不伴有血清IgG4升高。部分研究显示当眼眶炎性假瘤影像学表现为眶下神经增粗时,常提示IgG4-ROD可能性大[12-14],应该加做IgG和IgG4免疫组化染色,并结合临床表现和实验室检查作出正确诊断。
Mikuliczs病又称淋巴上皮病变,患者常表现为泪腺对称性、无痛性肿大,病理学表现为泪腺结节状增大,泪腺腺泡之间的基质内有大量成熟淋巴细胞弥漫性增生,伴有大小不一淋巴滤泡形成,泪腺腺泡萎缩,残存的泪腺小导管上皮增生,并形成许多界线清楚的上皮岛。Yamamoto等[15]研究发现Mikuliczs病属于一种IgG4相关性疾病,2011年Geyer等[16]建议采用IgG4相关性泪腺炎来定义泪腺Mikuliczs病,这一建议逐渐被接纳但仍存在诸多问题。基于2015年Goto等[5]提出的IgG4-ROD诊断标准,本实验也回顾性分析了15例Mikuliczs病,其中5例符合IgG4-ROD诊断标准归入IgG4-ROD组,其余10例中有5例出现散在IgG4阳性浆细胞但并未达到IgG4-ROD诊断标准,归入对照组。目前国际广泛使用的2015年Goto等[5]提出的IgG4-ROD诊断标准,大部分Mikuliczs病仍不能简单的定义为IgG4-ROD,特别是当患者不伴有血清IgG4水平升高时应谨慎诊断。
IgG4-ROD目前尚无有效统一的治疗方案,多采用局部手术切除及激素治疗,对于侵犯周围组织的患者往往无法完整切除,存在复发风险。目前多采用糖皮质激素治疗IgG4-ROD[17-22],大部分患者症状可以得到缓解,但约2/3患者停药或减量后复发,纤维化显著或血清IgG4水平低的患者对糖皮质激素治疗反应差[17]。对于伴发眼外受累的患者联合使用激素治疗和免疫抑制剂治疗可以显著降低患者的复发率[18]。IgG4-ROD是近年来才逐渐被认识的一类疾病,与眼部其他疾病在临床表现、影像学和病理特征上均存在一定相似性,对诊断造成困难,病理及临床医师应结合患者病史、实验室检查和免疫组化结果等综合考虑,必要时可借助分子检测进行鉴别诊断,并要积极排除其他部位是否存在系统性IgG4-RD,对确诊患者采取积极的治疗方案以降低其复发率。
