基于公共数据集的三阴性乳腺癌转录组学分析
2021-09-23龚俊杰徐子金
龚俊杰,王 平,徐子金
1)浙江工业大学药学院,浙江杭州310014;2)义乌市中心医院药剂科,浙江义乌322099
世界卫生组织国际癌症研究机构(international agency for research on cancer, IARC)发布的全球最新癌症负担数据表明,2020年全球乳腺癌新发病例高达226万例,超过了肺癌(220万例),成为全球第一大癌,发病形势日趋严峻.三阴性乳腺癌(triple negative breast cancer, TNBC)约占所有乳腺癌的15%,转移性TNBC患者的中位总生存期仅约为18个月.由于其病理学特征呈现雌激素受体(estrogen receptor, ER)、孕激素受体(progesterone receptor, PR)和人类表皮生长因子受体2(human epidermal growth factor receptor 2, HER-2)均为阴性,具有高度异质性和侵袭性,是最致命的乳腺癌亚型[1].目前,对TNBC仍缺乏有效的治疗手段,化学疗法是主要的辅助治疗方法,但易使患者产生耐药性[2].对ER阳性的乳腺癌,西达本胺能够逆转内分泌药物治疗的耐药性,从而提高他莫昔芬等的抗乳腺癌活性,但是,TNBC患者因ER检测结果为阴性,一般的化疗药物难以在此类乳腺癌中发挥作用[3],因此,TNBC的治疗手段十分受限,且疗效不佳.寻找与TNBC病变相关的生物标志物,并由此挖掘新型的TNBC诊疗靶标具有重要的临床应用意义.
转录组测序是了解特定条件下细胞中核糖核酸(ribonucleic acid, RNA)分子的类型和丰度的重要检测手段[4],现已经推广应用于多种RNA层面的基础与临床研究.比如,单细胞基因表达、RNA翻译和RNA结构组学等[5].基于转录组测序获得的表达谱数据进行生物信息学分析,旨在对复杂的高维数据进行分析,进而挖掘与疾病相关的关键基因和通路,揭示其在疾病发生和发展过程中基因的转录水平的变化规律,从而解析疾病所涉及的分子病理机制和诊疗靶标.
生物体系统的复杂性决定了转录组数据的高维性和庞大性.加权基因共表达网络分析(weighted gene coexpression network analysis, WGCNA)是除转录组常规分析外又一从复杂转录组数据中挖掘有效信息的重要方法,在疾病诊断和发病机制等生物医学研究中有广泛应用[6].基于表达模式相似的基因在功能上可能是紧密相关的以及生物体内蛋白质互作网络呈现无尺度化分布规律的两大基本生物学原理,通过WGCNA分析可鉴定表达模式高度相关的基因簇,提取与表型性状相关的基因共表达模块,将基因的表达模式与生物表型进行关联,有助于关注到与性状相关性较大的基因模块,构建基因模块的共表达网络,挖掘模块内关键基因.将该分析结果与转录组常规分析的结果进行比较或结合,可为关键基因的确立提供证据.
本研究利用公共基因表达谱(gene expression omnibus, GEO)数据库的TNBC相关基因子集GSE76275和GSE61724,借助转录组学分析方法,探究TNBC新型病理标志物,预测TNBC潜在的诊疗与干预靶标.
1 分析方法
1.1 数据来源
从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi)检索TNBC相关转录组表达谱基因集, 选择标准为: ① TNBC和非TNBC基因集;② 入组分析的样本未经药物干预和转基因等处理;③ 基因集对应的物种为智人(Homo sapiens);④ 基因集每组样本数至少为15个;⑤ 转录组数据分析集和验证集所使用芯片为同一家公司开发的芯片.最终选定GSE76275和GSE61724两套基因表达数据集作为本次转录组分析研究的数据.GSE76275数据集包含198个TNBC样本和67个非TNBC样本.非TNBC指除病理检测表型为三阴性以外的其他类型乳腺癌,本研究作为TNBC的对照样本.基因芯片型号是Affymetrix公司的人类基因组U133 plus 2.0芯片.另外,GSE61724数据集作为分析得到的关键基因的验证基因集,包含16个TNBC 样本和48个非TNBC样本.基因芯片型号是Affymetrix公司的人类基因组1.0 ST芯片.
1.2 主成分分析
利用R软件(x64 4.0.2)中的FactoMineR包实现主成分分析(principal component analysis,PCA).文中提到的R包均来自于Bioconductor数据库.
1.3 差异基因筛选
基因芯片数据运用R软件中的Limma包进行差异基因分析,差异表达基因分析筛选标准: lbF>1或lbF<-1,P′<0.05. 其中,F为差异倍数(fold change, FC),P′为校正后的P值. 本研究以TNBC为实验组(T),非TNBC为对照组(NT),比较实验组与对照组的基因表达水平的变化.
1.4 GO功能富集和KEGG通路富集
基因本体(gene ontology, GO)功能富集分析是一个在生物信息学领域中广泛使用的研究方法, 京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes, KEGG)是一个预测细胞中复杂信号通路的数据库,通过GO功能富集和KEGG通路富集,可获得与某一表型改变相关的病理生理功能的基因集合.因此,将差异基因与背景基因作比较,通过统计学方法识别显著富集的基因集合,能够在一定程度上了解是哪些基因功能或信号通路的改变可能涉及临床表型的改变,从而为疾病发生机制的分子调控提供研究思路.通过对Bioconductor数据库中的Clusterprofile包进行GO功能富集和KEGG通路富集, 当P<0.05时,认为富集具有显著性.
Metascape数据库(http://metascape.org)是一个功能强大的基因功能注释工具,可同时对GO和KEGG进行功能注释[7].在对差异表达基因和WGNCA共表达基因模块的交集基因的功能注释中,本研究使用Metascape数据库进行富集.以柱状图形式展示富集显著性排名前20的GO或KEGG条目,当-lgP>1.3时,认为富集是显著的.
1.5 蛋白质互作网络构建
利用STRING在线数据库(https://string-db.org/)构建蛋白质互作网络(protein-protein interaction,PPI),以预测蛋白质之间的相互作用关系,置信度设置为0.70,并统计排名靠前的节点度值.
利用R软件中的WGCNA包执行该分析方法.通过Cytoscape3.7.2软件构建基因共表达网络.
2 结果与分析
2.1 主成分分析
利用PCA方法评价GSE76275数据集样本组间和组内的分布情况,结果如图1.由图1可见,两组样本具有组内聚集,组间分散的分布特点,样本的辨识度较好,具有后续分析的价值.

图1 三阴性和非三阴性样本组主成分分析Fig.1 Principal component analysis of triple negative and non-triple negative sample groups

图2 差异表达基因火山图Fig.2 Volcano map of differentially expressed genes
2.2 差异表达基因的筛选
通过差异基因筛选共得到与TNBC病变相关的差异表达基因306个,如图2.其中,123个是上调基因,183个是下调基因.请扫描文末右下角二维码查看图S1,图S1给出了306个差异基因表达值热图的聚类分析,这些差异基因对TN和NTN两种乳腺癌病理表型具有较高的辨识度,提示部分基因表达模式的改变与乳腺癌不同病理分型的形成具有相关性.通过对这部分差异基因的富集分析,可为研究介导乳腺癌亚型病理分型上差异的潜在分子机制提供线索.
2.3 GO和KEGG富集分析
对306个在TNBC和非TNBC中差异表达的基因进行GO功能注释, 富集结果如图3, 图内颜色越深表示此GO 功能富集越显著,圆圈越大表示此GO 功能富集到的基因数越多.由图3可知,表皮细胞形成和分化、皮肤形成和皮肤角质化等生物学过程显著富集.顶端浆细胞膜、中间丝和着丝粒等相关细胞成分显著富集.转录激活因子结合活性、芳香化酶活性、N-乙酰氨基半乳糖转移酶活性、肌营养不良蛋白聚糖结合等分子功能富集显著.KEGG通路富集结果如图4,由图4可知,雌激素信号通路和过氧化物酶体增殖物激活受体(peroxisome proliferators-activated receptors, PPARs)信号通路被显著富集.以上的结果提示,这些功能或通路的改变可能和TNBC的快速增殖、侵袭转移等肿瘤的生物学特性相关.
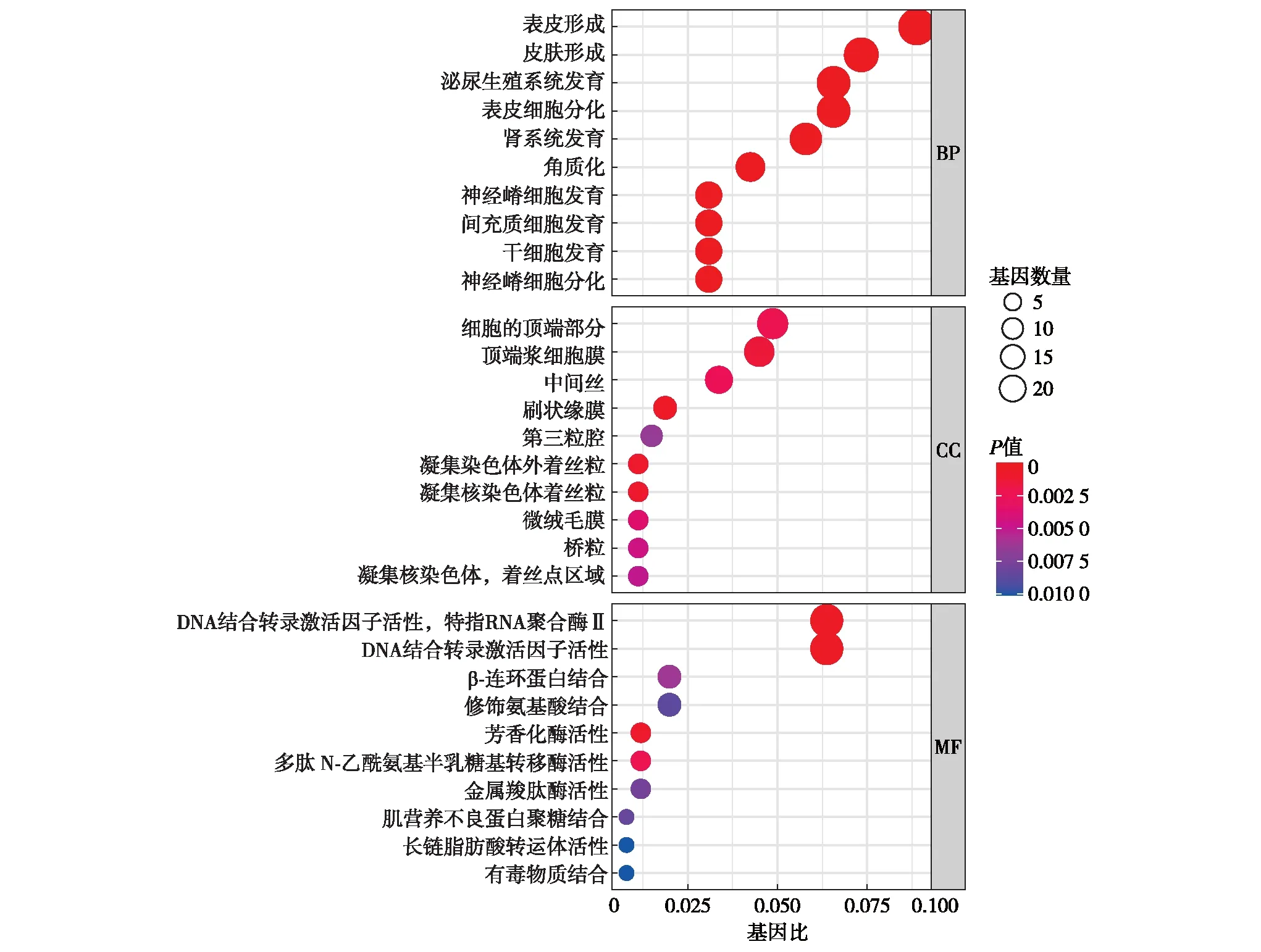
图3 GO功能注释Fig.3 GO functional annotation
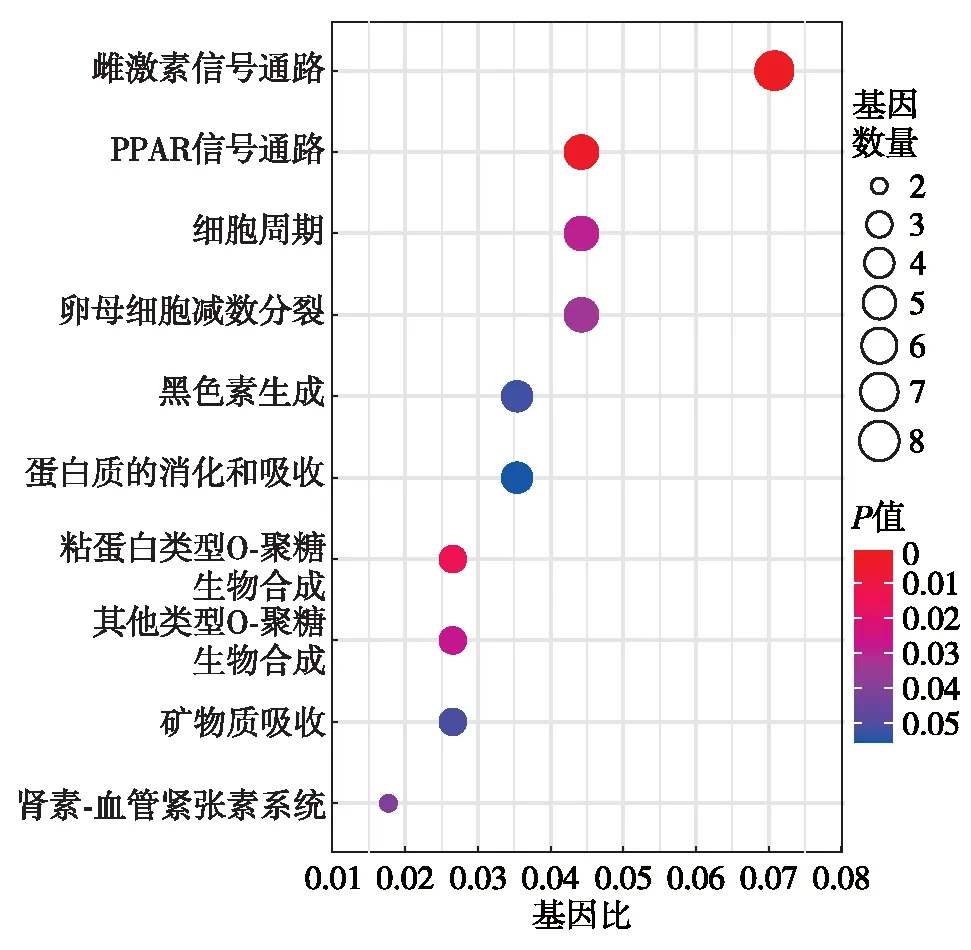
图4 KEGG通路富集分析Fig.4 KEGG pathway enrichment analysis
2.4 WGCNA分析
2.4.1 样本评价
基因芯片数据包含几万个探针对基因表达水平的检测结果,而其中表达水平显著变化的基因相比基因总数来说总是少数,为减少运算量和提高数据分析的效率,可以着重关注在所有基因中变异相对较大的基因[8],挑选GSE76275数据集中方差较大的前25%的基因(5 414个)表达谱进行后续的WGCNA分析.首先对265个样本进行初步层次聚类分析.剔除8个离群样本,样本的具体信息和样本层次聚类树结果请扫描文末右下角二维码查看补充材料图S2和图S3.
2.4.2 软阈值筛选
软阈值的作用是将基因间的强弱相关性放大,使基因共表达网络更符合无尺度化的分布规律,从而使其更贴近生物体系统的基因调控规律.最佳软阈值的筛选应同时满足:① 无标度拓朴模型拟合参数R2≥0.8; ② 不同模块中基因的平均连接度应比较高.综合考虑,本研究采用程序执行时自动筛选的6作为最佳软阈值(请扫描文末右下角二维码查看补充材料图S4),此时R2=0.87, 平均连通度为9.
2.4.3 基因模块层次聚类树
通过动态剪切树算法识别基因模块,模块内最少的基因数目设置为30,共有5 414个基因被分成了19个颜色不同的模块(请扫描文末右下角二维码查看补充材料图S5).将整个模块的基因表达谱通过主成分降维的方法进行特征提取,将表达谱的二维矩阵转化为一维向量,后者类似于模块内基因表达谱矩阵降维后的第1主成分,用module eigengenes (ME)表示,以抽象方式提取了模块内基因整体上的表达特征,据此计算相关性并进行聚类,合并相关性大于0.8的模块(请扫描文末右下角二维码查看补充材料图S6).合并后模块数减少至15个,不同颜色代表不同的基因模块,最大的模块是包含1 565个基因的紫色模块,最小的模块是仅有32个基因的浅绿色模块,如图5.
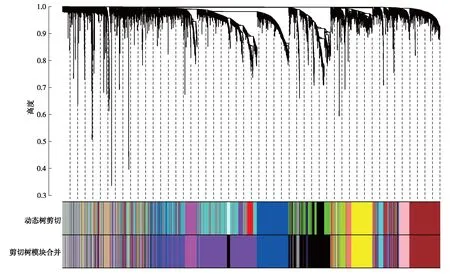
图5 相似基因模块合并Fig.5 Similar gene module merging
2.4.4 模块与表型关联分析
样本表型可转化为连续的数值型变量.基因作为沟通模块和表型的“桥梁”,计算模块与基因、表型与基因的相关性,继而得到模块-表型相关性热图,识别与表型显著相关的模块(图6).由图6可见,棕色模块与乳腺癌的三阴性表型相对有最显著的相关性(P<0.05). 故后续对棕色模块内的基因做进一步分析.
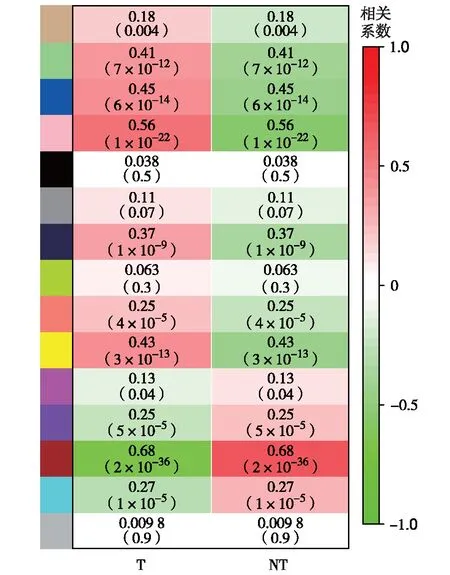
图6 模块-表型关联(括号内数值为P值)Fig.6 Module-phenotype association(The numbers in parentheses are P values.)
2.4.5 模块基因特征值与基因显著性的相关性分析

图7 棕色模块中MM-GS的相关性Fig.7 MM-GS correlation in brown module
模块内每一个基因与模块特征建立的相关性由 MM(module membership)值表示,MM值越大,表示模块内的基因与模块的相关性越大.GS(gene significance)值表示某基因与表型的相关性,该值越大的基因与表型越相关,该基因反映的生物学意义越大.因此,通过基因作为“桥梁”可将模块与表型的相关性联系起来.图7呈现的是棕色模块内所有基因对应的MM值和|GS|值,1个圆圈表示1个基因.两者呈较强的正相关性,相关系数达到0.78,一般地,图中越靠近右上角的基因与表型和模块的相关性越大,即这些基因在模块特征提取时的贡献度越大,提示这部分基因在乳腺癌的发生与发展过程中可能扮演更重要的角色.TANG等[8]把符合MM>0.8和|GS|>0.2的基因作为核心基因.基因与基因之间的表达具有相关性,即共表达关系,WGCNA通过取幂加权的方式更好的刻画了这种关系.图8是对棕色模块内权重相对较大的部分共表达基因对进行展示,网络中节点连线代表两基因间有共表达关系, 粗细代表权重的大小, 权重越大,基因间的相关性越大.模块内连接度(intramodular connectivity, IC)衡量给定基因相对于特定模块的基因的连接或共表达程度,该值越大表示某基因在共表达网络中的地位越重要,在网络中可能协同多个基因表达,并以此作为某一生物学功能或通路的关键调控基因. 图8为棕色模块的部分共表达网络,由图8可知,AGR3、MLPH、TBC1D9、AGR2、FOXA1和TFF1等基因具有较大的模块内连通度.分析可知,这些基因与所在的模块和所属的表型均具有显著相关性,其IC值、MM值和|GS| 值如表1,是WGCNA分析得到的重要基因.
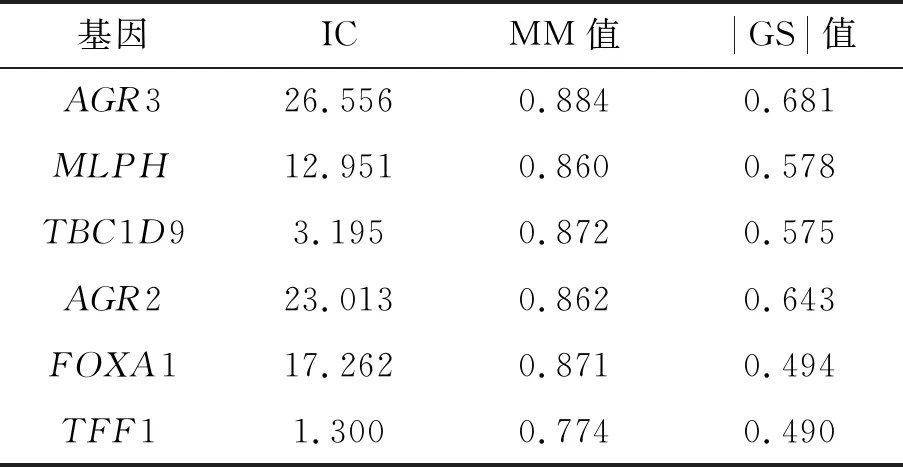
表1 棕色模块共表达基因的相关参数

图8 棕色模块的部分共表达网络Fig.8 A part of co-expression network in brown module
2.4.6 棕色模块的差异共表达基因分析
将棕色模块内的618个基因与306个差异基因取交集,得到141个差异的共表达基因,这些基因的表达特征不仅对乳腺癌的病理表型具有较好的辨识度,而且可能作为模块的共表达基因协同介导某种生物学功能或通路执行.通过Metascape数据库的富集分析表明,显著富集的基因本体是细胞内雌激素受体的正向调节和乳腺上皮细胞的生长发育,相关信号通路与2.3节富集结果一定程度上吻合,进一步提示乳腺癌的病理分型的形成可能涉及乳腺表皮或上皮细胞的增殖,以及性激素的相关调节等(图9).通过STRING数据库预测114个基因的蛋白相互作用关系网络,结果见图10.网络度值排名前12的节点见图11.一般认为,在蛋白互作网络中具有较大网络度值的基因为核心基因,这些基因可能对某一生物学功能的执行处于中心的调控地位.值得注意的是,结合前面的基因共表达网络分析的结果,本研究发现,三叶形因子1(trefoil factor1,TFF1)、叉头盒 A1转录因子(forkhead box A1,FOXA1)、人前梯度蛋白2(anterior gradient protein 2,AGR2)和人前梯度蛋白3(anterior gradient protein 3, AGR3)这4个蛋白不仅在蛋白互作网络度值较大而处于核心地位,而且在共表达网络中的模块内连通度较高,它们与模块、表型的相关性均较强,故作为本次转录组学分析输出的关键基因,后续作为重点分析和讨论的对象.在基因表达谱中获取它们在TNBC和非TNBC患者中的相对表达量,如图12(a)—(d)所示,发现4个基因在TNBC患者中的表达均显著地降低.并且,在另一个GEO数据库的基因集GSE61724这4个基因的表达模式也得到进一步确认,如图12(e)—(h).YI等[9]通过TNBC相关基因集分析发现,TNBC患者肿瘤组织中的TFF1表达比非TNBC患者肿瘤组织的表达低,病理学和血清学检测结果相同.这一发现与本研究分析结果一致,且血清TFF1水平与乳腺癌ER、PR和HER2的组织学类型存在显著关联.生存分析表明,TFF1的低表达与TNBC患者生存期减少显著相关.

图9 交集基因富集分析Fig.9 Enrichment analysis of intersection genes

图10 交集基因的蛋白互作网络Fig.10 Intersection genes of protein-protein interaction network
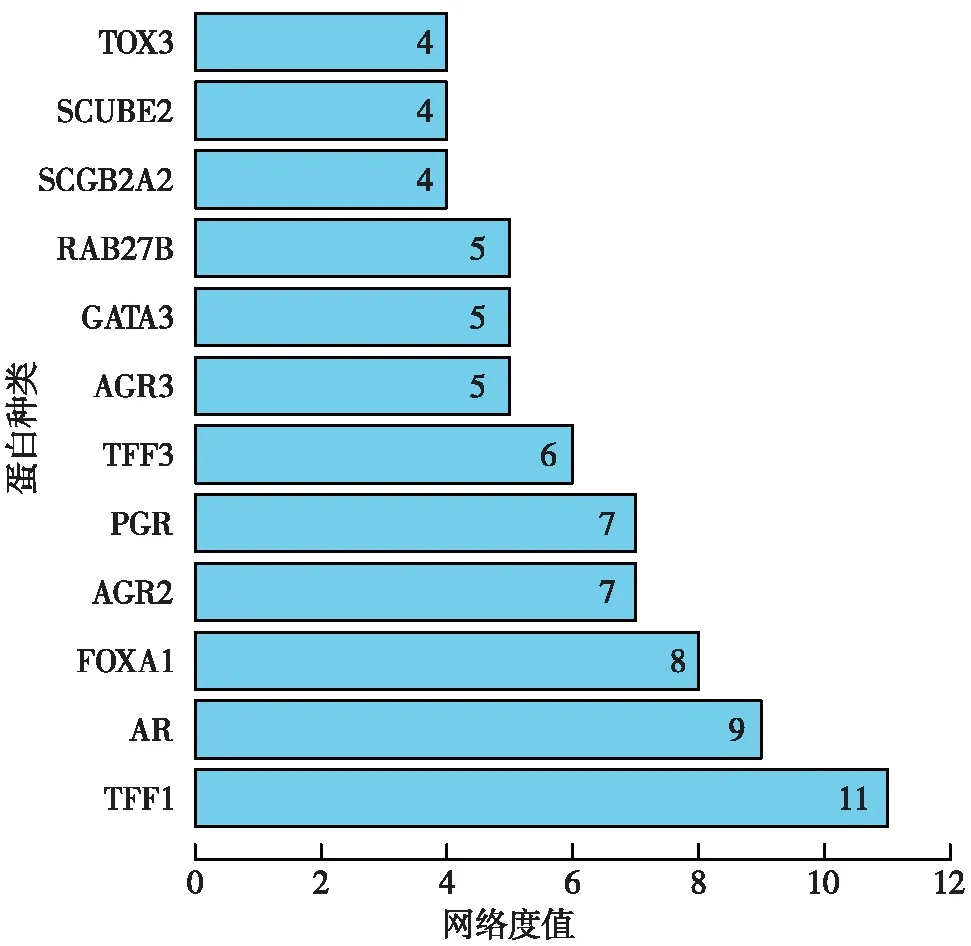
图11 蛋白互作网络度值统计Fig.11 Degree values of protein-protein interaction network with statistics
综上,TFF1的低表达可能与 TNBC的不良预后有一定相关性.因此,TFF1不仅可作为TNBC预后改善的潜在标志物,而且调控TFF1对于实现基于TNBC的分子分型的精准治疗可能具有较重要的意义.鉴于乳腺癌是一种与性激素内分泌水平高度相关的肿瘤,除了受到体内雌孕激素水平的调节外,雄激素环境对乳腺癌的发生和发展的相关研究则相对较少,而绝经女性体内的雄激素水平比雌孕激素水平的变化幅度小,提示雄激素在肿瘤的发生和发展过程中可能扮演着重要的角色[10].有研究指出,在TNBC中FOXA1与AR存在共表达关系[11-12].本研究特意关注了GSE76275数据集中的雄激素受体(androgen receptor, AR)与转录因子FOXA1的表达相关性,发现两者在表达水平上具有较强的正相关性,AR基因的表达随着FOXA1表达水平的升高而升高,两者存在一定的共表达关系,如图13.GUIU等[12]开展的队列研究发现有42%的非转移性TNBC表现为FOXA1 和AR共表达.AR 和 FOXA1阳性的TNBC其形态特征不同于其他亚型的TNBC,它是类似管腔肿瘤的TNBC亚型,该种亚型的TNBC预后结局较差,晚期复发风险较高.文献[13]认为,FOXA1发挥转录因子活性介导乳腺癌中AR驱动的转录,它作为先驱的协同转录因子与DNA结合,同时引导AR至通常由ER占据的结合位点,而AR通过依赖DNA结合的独立机制调节不同基因的分子功能(如ER的转录),从而诱导刺激细胞增殖的类似雌激素的基因程序.然而,也有研究认为,FOXA1是乳腺癌细胞的生长抑制剂和良好预后标志物[14].WU等[15]研究发现,FOXA1能够抑制TNBC细胞的上皮间质转化过程(epithelial-mesenchymal transition,EMT),从而阻止癌细胞的转移和侵袭.LIU等[16]发现FOXA1是促凋亡蛋白Bim的转录因子,抑制FOXA1的糖基化修饰,可增强FOXA1稳定性,从而促进乳腺癌细胞的调亡.另外,文献[17-18]研究均认为,FOXA1表达与乳腺癌良好预后和乳腺癌患者总体生存期延长相关.因此,FOXA1可能是鉴别TNBC特殊亚型和预后评价的生物标志物.GUO等[19]基于对公共数据库的乳腺癌基因集的分析,以及基因和蛋白层面的验证,发现TNBC组织中的AGR2表达明显低于非TNBC组织,与本研究结果一致.同时,经统计学检验表明,AGR2 表达与组织学类型、组织学分级、ER 状态和 PR 状态显著相关.临床病理变量的单变量分析表明,AGR2在乳腺癌中的高表达与总体生存期较短显著相关.Cox 多变量分析认为,AGR2可以作为一个独立的关于肿瘤不利结果的候选指示因子[19],因此,AGR2的表达水平对于乳腺癌的病理分型具有一定的区分能力,AGR2可能在乳腺癌诊断和预后中具有重要的价值.AGR3和AGR2均属于二硫化物异构酶家族,具有高度的序列同源性,因此它们可能介导了肿瘤生物学的相似方面.AGR2已被广泛证明与肿瘤的进展有关,但对AGR3在肿瘤生物学中的作用却了解较少[20].OBACZ等[21]评估了AGR3在乳腺癌患者中的临床和预后意义,发现AGR3阳性细胞百分比与ER、PR状态以及肿瘤低组织学分级显著相关,AGR3的表达与肿瘤增殖标志物Ki-67表达水平呈显著负相关,AGR3 阳性亚组的低组织学分级肿瘤显示出更长的无进展生存期和总生存期,AGR3与乳腺癌患者有利的预后有关.然而,一些研究也认为AGR3具有致癌潜能,具体表现为它可能和乳腺癌细胞的增殖、迁移以及耐药等肿瘤生物学特性相关[22-24].

图12 基因相对表达量Fig.12 Relative expression of genes
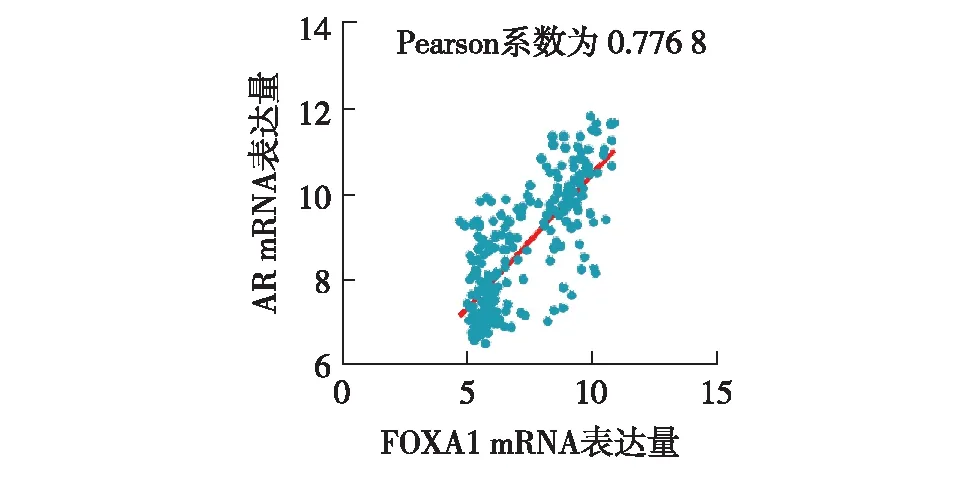
图13 FOXA1和AR表达量的相关性Fig.13 Correlation between FOXA1 and AR expression levels
结 语
TNBC的治疗目前依然面临着可干预靶点少和治疗药物选择性小的困境.本研究基于转录组学方法对公共数据库的基因集数据分析发现,TNBC与非TNBC在基因表达、富集的生物功能和信号通路上存在着明显的差异.TFF1、FOXA1、AGR2和AGR3基因有可能作为TNBC临床表型辅助诊断与鉴别诊断,以及精准治疗与预后干预的关键分子靶标,值得深入研究和进一步验证.
