慢性社交挫败应激小鼠大脑的C-U RNA编辑
2021-09-23任春艳魏志远饶军华饶义剑陈建欢
任春艳,魏志远,饶军华,饶义剑,陈建欢
1)江南大学无锡医学院,江苏无锡 214122;2)江南大学生物工程学院,江苏无锡214122;3)江南大学-广东科学院动物研究所“慢性病灵长类研究联合实验室”,江苏无锡214122;4)广东科学院动物研究所,广东广州510260
核糖核酸(ribonucleic acid, RNA)编辑是在转录水平上发生的RNA核苷酸序列的改变[1].经典RNA编辑主要包括由ADAR(adenosine deaminases acting on RNA)蛋白家族介导的腺苷到肌苷(A-I)和APOBEC(apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like)蛋白家族介导的胞苷到尿苷(C-U)的编辑.RNA编辑目前已在动物、植物和人等多种生物中被发现,并参与多种生理及病理学过程.近期研究提示,RNA编辑在应激、神经系统疾病和精神病理性疾病条件下存在明显变化[2-3],提示其可能参与这些过程或疾病的分子机制.
社交挫败模型主要用于研究创伤后应激障碍(post-traumatic stress disorder,PTSD)、抑郁症和其他应激相关的疾病.文献[4]研究表明,社交挫败应激可诱导RNA编辑的变化.DICK等[5]的研究提示,小鼠在慢性社交挫败应激(chronic social defeat stress, CSDS)后,其大脑内侧前额叶皮层和基底外侧杏仁核内存在A-I编辑的动态调节.但是C-U的RNA编辑在情绪应激模型中的变化如何,目前尚不明确.有意思的是,单纯通过目击创伤性事件所引起的情绪应激(emotional stress, PS)与通过身体接触所引起的生理应激(physical stress, ES)诱导的小鼠大脑转录组表达谱存在差异,因此,不同应激条件下CSDS小鼠模型中C-U的RNA编辑尚待研究.
本研究通过RNA测序,在转录组范围内识别大脑的腹侧被盖区(ventral tegmental area, VTA)中的C-U RNA编辑,研究其分布情况.通过比较不同条件下的CSDS过程中C-U RNA编辑情况,分析其对基因功能和通路的潜在影响,以及参与调控的潜在分子机制.
1 材料与方法
1.1 RNA测序数据的获取
RNA测序原始数据从美国国家生物信息中心(National Center for Biotechnology Information, NCBI)的Gene Expression Omnibus(GEO)数据库中(https://www.ncbi.nlm.nih.gov/geo/)下载获得.ES组、PS组以及对照组(每组设平行实验N=3)的成年雄性小鼠大脑腹侧被盖区(ventral tegmental area, VTA)RNA测序数据(GSE36005)[4].RNA编辑位点的验证数据包含44个成年小鼠大脑VTA RNA测序(GSE89692)[6-7].
1.2 RNA测序数据的比对
利用FASTQC软件对以上获得的原始测序数据进行质量控制分析.质控合格的测序读段(reads),利用RNA STAR (2.7.0e版)[8]软件与加利福尼亚大学圣克鲁兹分校(University of California, Santa Cruz, UCSC)小鼠的基因组序列(mm10版)进行比对,分析其RNA剪接接头,将其定位(map)到基因组上,并生成二进制比对映射(binary alignment map, BAM)格式的比对结果文件.利用Samtools(1.9版)软件将BAM文件中的读段去重复(deduplication)[9],只保留有单一定位的读段,并使用GATK(4.1.3版)软件对BAM文件进行碱基质量评分重校准[10].
1.3 RNA编辑位点的鉴定及注释
利用VarScan(2.4.3版)软件对校准处理后的BAM文件进行RNA单核苷酸变异(single nucleotide variant, SNV)识别[11].识别标准为碱基质量≥25,测序深度≥10,U碱基(RNA测序中被转换为T碱基)深度≥2且频率≥1%,并用VarScan的 fpfilter命令和默认参数过滤假阳性变异.对达到以下任一条件的SNV均予以滤除[12]:① 位于简单重复序列或5个连续相同碱基内;② 位于线粒体内;③ 距离RNA剪接接头6个核苷酸以内;④ 距离插入或删除变异1个核苷酸以内;⑤ 在dbSNP数据库中的已知变异相吻合.为进一步提高RNA编辑位点识别的可信度,只保留在样本或验证数据样本中都被检测到的,即可重复验证的编辑水平≥1%的C-T SNV,将其定义为高置信度变异.SNV的注释使用Ensembl Variant Effect Predictor (VEP, https://www.ensembl.org/vep)软件进行[13].
1.4 基因表达的定量及差异分析
采用FeatureCounts软件对RNA STAR生成的比对文件进行分析,获得RNA表达的计数,并用EdgeR软件(3.7版)计算标准化的基因表达水平(transcripts per kilobase million,TPM)[14].
1.5 主成分分析
利用1.4节获得的基因表达的定量数据,使用R软件(3.6.3版)中的函数Prcomp进行主成分分析(principal component analysis, PCA),并使用ggplot2(2.2.1版)软件包进行数据可视化.
1.6 基因功能和通路的富集分析
为了理解RNA编辑的潜在生物学效应,利用Enrichr软件(https://maayanlab.cloud/Enrichr/)[15]分析发生RNA编辑的基因所富集的gene ontology(GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路等,以错误发现率(false discovery rate, FDR) < 0.05作为筛选条件.
1.7 统计学分析
使用Kruskal-Wallis(KW)非参数检验,比较样本组间RNA编辑水平或基因表达水平.组间的两两比较用Dunn校验进行,并用Benjamini-Hochberg法对多重比较的P值进行校正.以P<0.05作为显著差异筛选标准.采用Spearman相关分析计算编辑位点间的相关系数R. 在有多个编辑位点的基因中,差异位点的富集分析使用Fisher精确检验进行.
2 结果与分析
2.1 小鼠VTA的C-U RNA 编辑
在3组成年小鼠VTA的RNA-Seq中,共在5 866个基因中发现了16 198个高置信度C-U RNA编辑位点,见图1(C-U RNA编辑位点的具体信息请扫描论文末页右下角二维码查看表S1).这些位点广泛存在于各染色体中,其样本间最高的RNA编辑频率为1%~100%.从每个基因的编辑位点数量来看,部分基因位点密度较亮,这些位点涵盖了多种功能类型,按数量多少依次为3′-非编辑区(untranslated region, UTR)变异(5 616个)、错义变异(4 953个)、同义变异(4 172个)、无义变异(716个)、5′-UTR变异(594个)和非编码转录本的外显子变异(147个), 如图2.

图1 成年小鼠VTA转录组中基因表达(外圈)、C-U RNA编辑位点(中圈)和位点间相关性的Circos图Fig.1 Circos plot showing the gene expression (outer circle), distribution of C-U RNA editing events (middle circle)and their correlation in adult mouse VTA
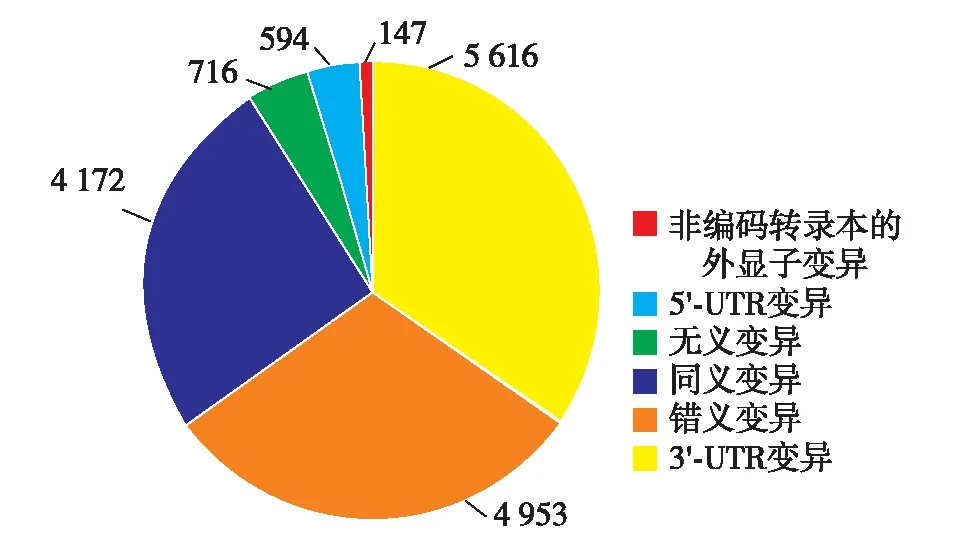
图2 成年小鼠VTA转录组中C-U RNA编辑SNV的功能类型Fig.2 Functional categories of C-U RNA editing variants in the mouse VTA
由图2可见,部分基因检测到多个编辑位点.编辑位点数量排前10位的基因请扫描论文末页右下角二维码查看表S2.

图3 48个位点在组间存在显著差异RNA编辑(所有位点的KW检验均有P<0.05)Fig.3 Forty-eight sites with significant differential RNA editing among the three groups RNA editing(all KW test P<0.05)
2.2 不同应激条件下的差异C-U RNA编辑
为分析不同应激条件下RNA编辑的改变,通过KW检验比较位点在不同分组间的RNA编辑水平,发现共有48个基因中的48个位点存在显著差异(图3).这些基因/位点有22个在ES中, 9个在PS组中,15个在对照组中有显著较高的编辑水平,另有2个在ES中有较低的编辑水平.PCA结果表明,这些差异编辑可以很好地将3组小鼠分开,其中,PC1和PC2的贡献率分别为51.78%和30.74%(图4).Fisher检验分析发现,这48个呈差异编辑的基因中44个(91.7%)具有1个以上的编辑位点,显示差异编辑显著富集于具多个编辑位点的基因中(优势比=8.5,95%置信区间为3.1~32.5,Fisher检验P=1.74×10-7).其中,差异C-U RNA编辑位点在有多个编辑位点基因中的富集情况请扫描论文末页右下角二维码查看表S3.
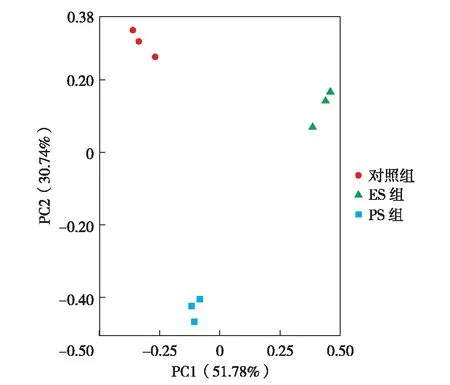
图4 48个差异编辑位点的主成分分析Fig.4 PCA analysis using forty-eight sites with differential RNA editing
2.3 RNA编辑的功能富集
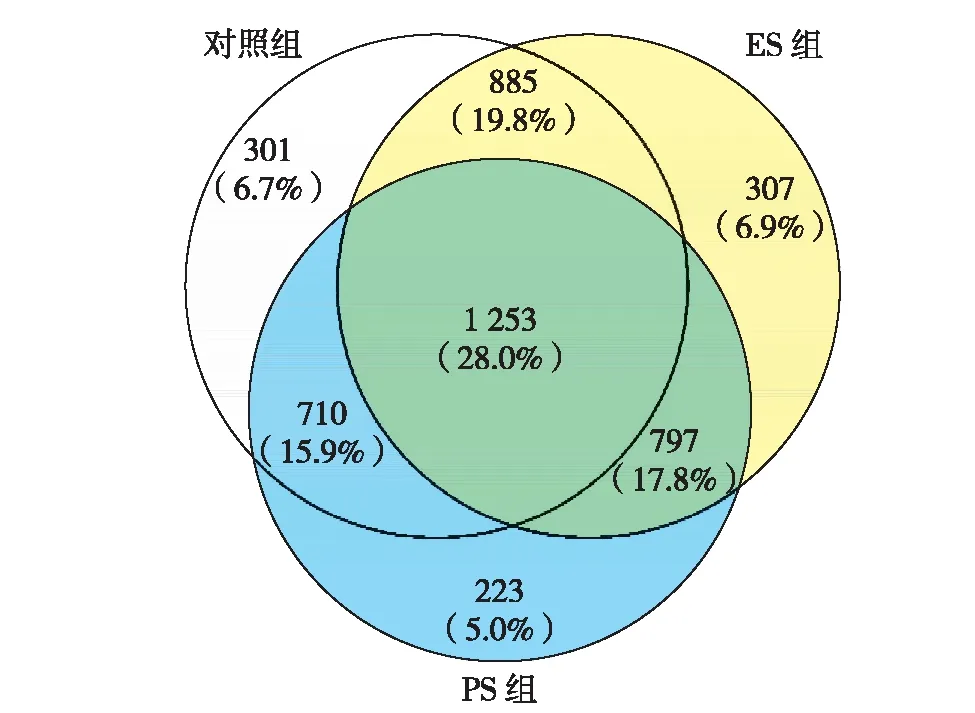
图5 各组中在2个或以上样本中检测到的C-U RNA编辑位点比较的维恩图Fig.5 Venn plot comparing the C-U editing sites detected in two or more samples among groups
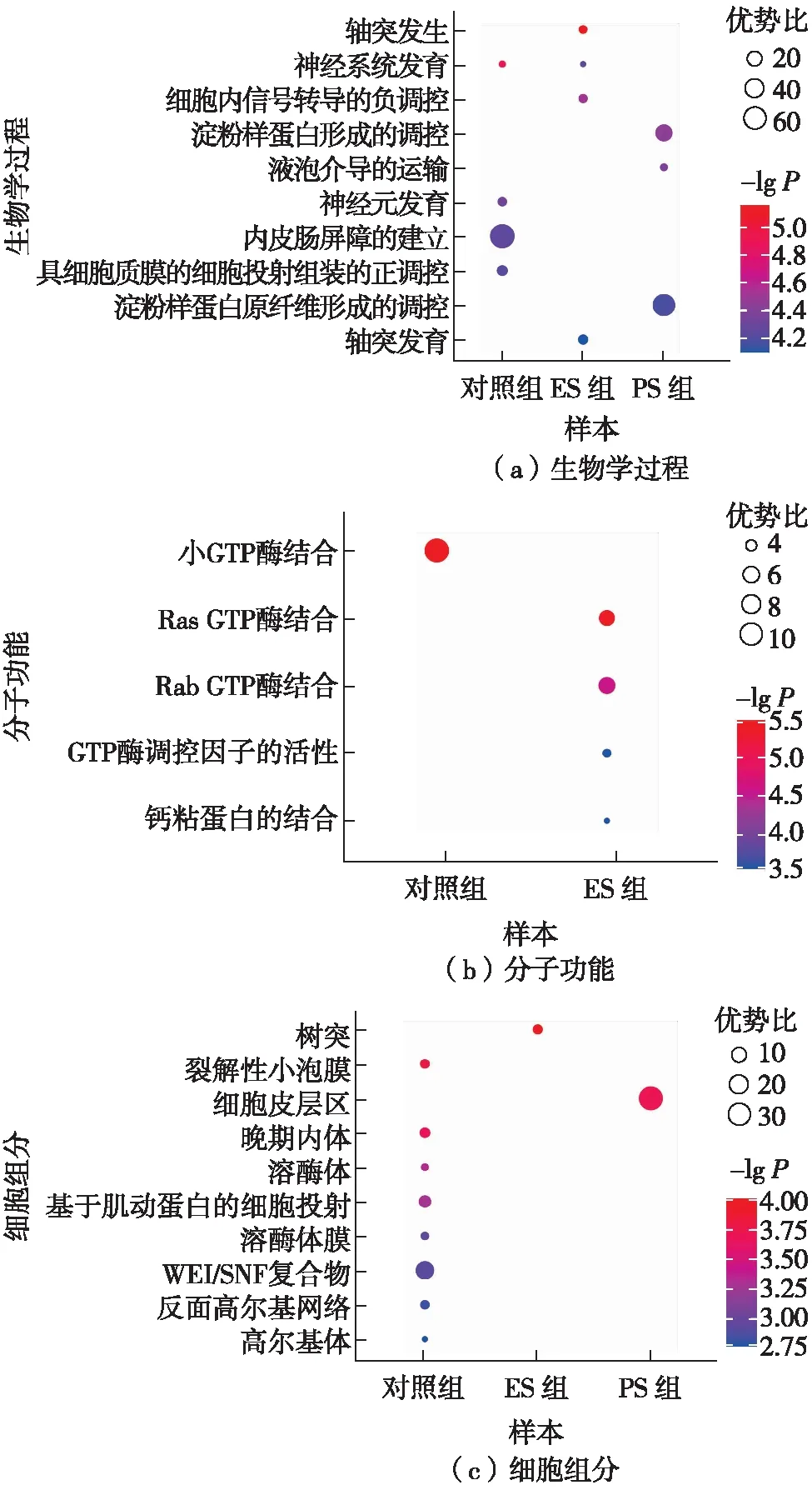
图6 GO分析显示各组独有的编辑位点的基因呈现出差异化的基因功能富集Fig.6 GO analysis showing differential enrichment of gene function in genes with editing sites unique to groups
进一步比较不同组中RNA编辑的存在情况,发现ES、PS和对照组中分别有307、223和301个编辑位点特异地存在于组内至少2个样本中,而在另外2组未检测到(图5).Enrichr基因列表富集分析结果表明,这些各组独有的RNA编辑呈现出不同的基因功能特点(图6).ES组特有的RNA编辑主要富集在轴突发生、神经系统发育、细胞内信号转导的负调控和轴突发育等生物学过程.这些基因主要参与Ras(rat sarcoma)等多种GTP(guanosine-5′-triphosphate)酶的结合活性、GTP酶调控因子的活性和钙黏蛋白的结合活性等分子功能,与树突等细胞组分有关.PS组独有的RNA编辑主要富集在淀粉样蛋白及其原纤维形成的调控和液泡介导运输等生物学过程,与细胞皮层区的细胞组分有关.而对照组中独有的RNA编辑主要富集在神经系统发育、神经元发育和具细胞质膜的细胞投射组装的正调控等生物学过程.这些基因主要参与小GTP酶结合活性等分子功能,与裂解性小泡膜、晚期内体、溶酶体、溶酶体膜、基于肌动蛋白的细胞投射、SWI/SNF(SWItch/sucrose non-fermentable)复合物、高尔基体和反面高尔基网络等细胞组分有关.
2.4 ES组中血清和糖皮质激素依赖性激酶1基因的改变
通过比较差异表达的基因和特异编辑位点发现,与对照相比,血清和糖皮质激素依赖性激酶1(serum/glucocorticoid regulated kinase 1,Sgk1)基因是ES组表达差异最为显著上调的基因之一,而在PS组中改变不明显(图7(a)),同时,在10号染色体的第21 999 752位点的3′-UTR位置,Sgk1基因也仅在ES组中有特异性编辑(图7(b)).
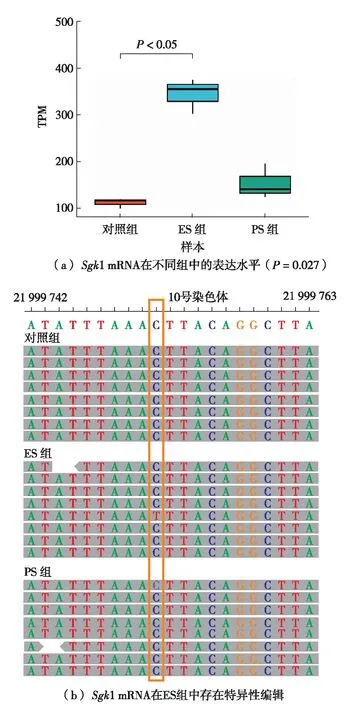
图7 小鼠VTA中Sgk1 mRNA的表达存在显著的组间差异及其在ES组中的特异性C-U RNA编辑Fig.7 Significantly differential mRNA expression and ES-specific C-U RNA editing of Sgk1 mRNA in the mouse VTA
3 讨 论
CSDS的啮齿类模型被广泛用于抑郁症等相关疾病的研究,为理解这部分疾病提供了重要的科学参考和依据.本研究首次在转录组范围内对CSDS小鼠模型中大脑VTA内存在的C-U RNA编辑进行了系统性研究.
VTA是大脑奖赏系统的关键区域,与应激相关行为关系密切[16].因此,在VTA中广泛存在的C-U RNA编辑可能与VTA的重要生物学功能有密切关系.这些编辑位点多集中于RNA的3′-UTR ,这与现有的C-U RNA编辑的研究结果一致.这些RNA编辑可能通过影响微小RNA与3′-UTR 的结合,从而调控转录后RNA的稳定性.此外,有研究还提示3′-UTR RNA编辑可能影响RNA翻译,从而作用于蛋白水平[17].C-U RNA编辑在不同基因间的分布并不均匀,部分基因存在数量较多的编辑位点,可能是编辑热点.编辑位点数量最多的10个基因多与中枢神经系统的功能及精神疾病有关,可能参与相关的生物学过程.例如,在ATP酶 Na+/K+转运亚其Atp1a3基因发现了47个C-U RNA编辑位点.ATP1A3基因编码神经元特异性ATP依赖的跨膜钠钾泵的α亚基,有研究报导ATP1A3的错义变异与儿童期精神分裂症相关[18].此外,CSDS差异编辑基因91%以上集中于这些有1个以上编辑位点的基因中,提示这些编辑热点所在的基因与CSDS之间存在潜在的重要联系.与之相关的是,小胶质细胞中C-U编辑功能的缺陷会加剧与年龄相关的中枢神经系统病理生理学改变[19].值得注意的是,已发现的RNA编辑变异中,编码变异占了相当大的比例(56.3%),而编码变异中的错义和无义变异加起来占编码变异的54.3%.这可能意味着C-U RNA编辑可能对VTA中表达的蛋白序列有潜在影响.这种影响的结果及其产生的原因是什么,尚待进一步研究.
与已报导的CSDS小鼠VTA中的A-I位点相比,C-U RNA编辑位点编辑水平普遍较低[5].这可能意味着与A>I RNA编辑相比,从RNA测序数据中识别C-U RNA编辑可能面临的难度更大.本研究中采用独立样本进行编辑位点的验证,并针对多等位基因位点进行了处理,从而提高了分析流程的可靠性.
在CSDS小鼠模型中,发现48个基因中的48个位点的编辑水平有显著的组间差异.同时PCA结果显示,这些位点PC1和PC2加起来可以解释超过81%的样本间变异.这些结果提示在ES、PS和对照组的中存在较明显的差异编辑.这些存在差异编辑水平的基因部分为已知的精神类疾病易感基因,如谷氨酸离子受体GRIA4被报导是尼古丁依赖和重度抑郁共同的风险基因[20];也有部分为编码RNA结合蛋白,如ELAV样RNA结合蛋白1 I(Elavl1)基因编码的Hu抗原R,能结合mRNA的3′-UTR并增加其稳定性,并与RNA编辑的功能调节相关[21].
谷氨酸受体已知是A>I RNA编辑重要的靶基因[22].本研究分析结果显示,离子型谷氨酸受体Gria4(glutamate ionotropic receptor AMPA type subunit 4)中存在显著的差异C-U RNA,提示谷氨酸受体相关基因可能也是重要靶点,并与应激反应相关.
特别重要的是,在这些特异性位点中, ES组中存在Sgk1基因同时存在特异性RNA编辑和表达水平改变.Sgk1编码的血清和糖皮质激素调节激酶1属于丝氨酸/苏氨酸蛋白激酶的一种,在应激和抑郁症等疾病中起重要作用[23].在慢性、轻度、不可预见应激或生命早期应激诱导的抑郁症大鼠模型中,海马的Sgk1 mRNA水平显着增加[24].在未用药抑郁症患者的外周血中同样检测到Sgk1基因表达量增加[24].首次发现的与ES相关的Sgk1 mRNA可能与其表达水平的增加有关,具体生物学功能有待进一步研究.
结 语
通过首次识别成年雄性小鼠大脑VTA转录组中存在的C-U RNA编辑,将不同CSDS模型与对照进行对比.结果揭示了CSDS模型特异性的C-U RNA编辑改变,为进一步理解RNA编辑参与CSDS的分子机制提供了依据.
