酱卤肉制品中食品添加剂的固相提取与精确分析
2021-09-21王晓君李凤玲
王晓君 李凤玲
(1. 烟台理工学院食品与生物工程系,山东 烟台 264005;2. 山东理工大学农业工程与食品科学学院,山东 淄博 255000)
食品添加剂在改善肉制品的色、香、味、形方面起着关键作用,对产品质量提升和降低生产成本也有一定的帮助,可以说食品添加剂有效地推动了肉类食品的高速发展[1-2]。根据国家食品安全标准规则,卤制肉制品中常用的添加剂按照用途划分有防腐剂、护色剂、抗氧化剂、着色剂、乳化剂等[3]。GB 2760—2014《食品安全国家标准 食品添加剂使用标准》对苯甲酸、山梨酸和糖精钠在肉制品加工过程中的添加量有严格的限定。但不法商贩为了延长产品的保存期限、保持肉制品的外观与气味,超量使用或违规使用添加剂的现象时有发生,严重危害消费者的利益及身心健康[4-6]。目前,GB 5009.28—2016《食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定》规定的测定方法有液相色谱法和气相色谱法两种。其中液相分析方法中对于高油脂试样的处理,经正己烷脱脂后采用亚铁氰化钾沉淀剂沉淀蛋白,分离过程过于简单,样品中存在油脂残留以及杂质的可能,无特异性吸附前处理,若直接使用C18柱,峰型特征无法保证。该方法各添加剂的保留时间较长,液相单针分析时间达到了18 min,分析效率不高,溶剂损耗大,且使用的沉淀剂亚铁氰化钾溶液易产生剧毒氰化物,后处理困难。由于肉制品含有大量的脂肪,传统的静置离心净化方法很难将肉制品中的脂肪直接去掉,残留的脂肪直接进样容易污染色谱柱[7-8]。厉建军等[9]曾用高效液相色谱分离肉制品中的添加剂,肉制品捣碎后需经超声处理、亚铁氰化钾溶液沉淀、离心才能检测,该方法的检测限仅为0.001 8 g/kg,加标回收率为85%~101%,难以满足日益增长的检测精密度需求。而且高效液相色谱法在对营养丰富的肉制品中的苯甲酸、山梨酸等防腐剂和糖精钠甜味剂进行检验时,常常会因保留时间的漂移和检测精密度问题影响到检测结果的判定[10]。尽管众多的研究开展了对添加剂的检测增加了不确定度分析,将主要原因归集为样品提取过程和仪器的精密度问题,拟通过加强操作人员的熟练度、规范试验操作,减少人为因素对提取过程的影响和使用精密度更高的仪器从而提高检测结果的准确性[11]。但人为的干预以及仪器的精密度无法解决复杂样品的杂质干扰和分析条件的选择性问题。研究拟选取特异性吸附的SPE-MCX固相萃取剂对成分复杂的酱卤制肉样品进行处理,结合HPLC法对添加剂进行定量分析并进行方法学验证,以期为酱卤制肉品中食品添加剂的检测提供参考。
1 试验部分
1.1 仪器与试剂
安捷伦高效液相色谱:LC-1260Ⅱ型,美国安捷伦公司;
液相色谱柱:Agilent ZORBAX Eclipse XDB-C18型,美国安捷伦公司;
可变速高速旋转粉碎机:PULVERISETTE 14型,北京飞驰科学仪器有限公司;
超声波清洗机:BKE-1004HT型,杭州博可超声波设备有限公司;
电子分析天平:BL610型,瑞士梅特勒公司;
高速冷冻离心机:TJ-140型,美国THERMO公司;
高真空旋转蒸发仪:R-300型,瑞士BUCHI公司;
苯甲酸、山梨酸、糖精钠标准品:纯度>98%,国家标准物中心;
正己烷、乙腈:色谱纯,德国默克公司;
甲醇、甲酸:分析纯,麦克林试剂公司;
1.2 磨损颗粒的制备 将真空球磨仪 (GN-Z高能球磨仪,沈阳市新科仪器机电设备厂)所有的组件按照ASTM F1903标准进行清洗,然后置于75%的酒精中浸泡,高温灭菌以备用。将磨块置入球磨罐中,加入100ml无水乙醇作为保护液,抽真空,开始球磨,每2h需要停机15min,以避免球磨罐温度过高发热。用400目筛网过滤乙醇和磨损颗粒,等待乙醇完全挥发后收集颗粒,本研究将球磨了12h、24h、48h、60h、72h 后获得的颗粒分别进行收集[9]。
SPE-MCX、SPE.WCX、SPE.SIL固相萃取剂:山东博纳生物集团;
五香脱骨卤猪蹄:购于重庆、本溪、青岛三地某食品公司。
1.2 溶液配制
1.2.1 样品前处理 切取5.0 g卤肉样品,加入5.0 g蒸馏水,置于粉碎机中,捣碎5 min形成肉泥,将破碎后的卤肉液体加5.0 mL乙酸乙酯,置于超声波清洗器中超声10 min,再8 000 r/min离心10 min,取5.0 mL上清液,置于50.0 mL容量瓶中用甲醇定容,并用冰醋酸调pH至3.5~4.0,摇匀。依次流加5.0 mL乙醇和5.0 mL超纯水在固相萃取柱中进行充分活化。然后移取5.0 mL上清液过柱,先用5.0 mL纯化水洗杂,再用70%甲醇溶液5.0 mL洗脱,速度均为1 BV/h,洗脱液收集于容量瓶中。经过0.45 μm有机滤膜过滤后,收集待检[12]。
1.2.2 标准溶液配制 分别称取各标准品适量,用纯化水超声10 min,充分溶解;置于100 mL棕色容量瓶中,配成浓度为100 mg/L标准品母液,备用。使用时,3种物质的母液分别取0.5,1.0,2.0,5.0,10.0 mL,各加入1 mL冰醋酸酸化并用高纯水定容至100 mL,制得相应浓度的标准溶液,用锡箔纸包裹,于4 ℃避光保存。
1.3 液相条件
Agilent ZORBAX Eclipse XDB-C18(250 mm×4.6 mm,5 μm);柱温25 ℃;流速0.8 mL/min;进样量10 μL;检测波长230 nm;流动相A为0.02 mol/L乙酸铵水溶液,B为乙腈,VA∶VB=75∶25;光谱全波长扫描范围210~400 nm。
1.4 试验设计
1.4.1 固相萃取优化 分别选取固相萃取剂包括强阳离子交换的SPE-MCX、弱阳离子交换的SPE.WCX和中强度离子交换的SPE.SIL对样品进行选择性吸附,选取最佳的固相萃取剂。然后配置浓度为5 g/L各待测成分标准溶液,分别用冰醋酸调整pH至2.0,3.5,7.0,进料速度为1 BV/h,通过分析各物质出料浓度与进料浓度的比值,考察pH的变化对苯甲酸、山梨酸、糖精钠在SPE-MCX固相萃取柱吸附效率的影响。在确定最佳的进料pH后,分别取5 mL含50 mg/L的苯甲酸、山梨酸和糖精钠上样,进料速度为1 BV/h,然后分别用5 mL纯水、含30%,60%,90%甲醇溶液各5 mL来洗脱,分别收集并测定洗脱液中苯甲酸、山梨酸和糖精钠含量,然后计算各组分的回收率。
1.4.2 专属性与保留时间优化 分别配制0.1 g/L苯甲酸、山梨酸和糖精钠溶液,使用DAD检测器在210~400 nm紫外全波长扫描,确定最佳的检测波长。在该吸收波长下,分别试验了3种食品添加剂在不同比例乙酸铵—乙腈溶液时的分离效果,流动相V乙酸铵∶V乙腈分别为90∶10,80∶20,72∶25,50∶50下的各物质保留时间。
1.4.3 方法学确认
(1) 线性范围与检测限:通过配置不同质量浓度的0.5,1.0,2.0,5.0,10.0 mg/L标准溶液进样得到的标准曲线来评估方法的线性范围及其相关性。以信噪比S/N=3计算检出限。
(2) 回收率与精密度:为了满足不同浓度下3种添加剂的检测的需求,分别在0.05,0.50,1.00 mg/kg 3个水平下进行加标回收试验。
2 结果与讨论
2.1 固相萃取吸附剂选择
如图1所示,总体来看,3种添加剂在固相吸附剂上的吸附能力为山梨酸>苯甲酸>糖精钠。同时,不同的吸附剂其阳离子强度越大,其吸附效果相对更高,表现为SPE-MCX>SPE.WCX>SPE.SIL。原因为SPE.SIL为二氧化硅基质的固相萃取剂主要用于非极性溶剂中脂肪酸类物质的吸附。而SPE-MCX和SPE.WCX主体基质为N-乙烯基吡咯烷酮—二乙烯基苯共聚物基质对极性分析物有高度的选择性。SPE-MCX连接有磺酸基团相对SPE.WCX连接的醋酸基团有更强的耐有机溶剂的稳定性。吸附的3种食品添加剂均为极性物质,因此,选取SPE-MCX固相萃取剂为后续的研究对象。

图1 不同添加剂在不同吸附剂的吸附量
2.2 固相萃取条件的选择
2.2.1 pH优化 SPE-MCX柱的吸附原理为目标化合物遇到酸化后呈阳离子特征,通过电荷以及分子间力结合,保留在小柱上,通过改变离子强度,pH值和破坏分子间力后洗脱下来。由图2可知,pH中性条件下,各组分在SPE-MCX固相上几乎不吸附,而随着pH值的降低,吸附量随之增大。当pH为3.5时,控制出料浓度<5%的进料浓度,各物质最佳的吸附量为8 BV。当进一步降低进料pH时,各物质在SPE-MCX固相上的吸附率出现不同程度的降低,考虑到SPE-MCX柱在pH<2时会发生降解的风险,所以选取进料pH为3.5左右作为后续的研究。

图2 吸附液pH对吸附效果的影响
2.2.2 洗脱液浓度优化 洗脱溶剂的选择直接影响产品的纯度和回收率,为了达到产品的高纯度,通常在洗脱之前增加洗杂步骤以保证产品的纯度,但是提取高纯度的产品不可避免地带来产品的损失,适合的洗脱溶剂浓度,成为洗脱收率的关键。由图3可知,以纯水洗脱时只有极少量的苯甲酸、山梨酸和糖精钠被洗脱下来,含一定量的甲醇的混合体系有利于产品的洗脱。当用体积分数60%的甲醇溶液洗脱时,使用3 BV的洗脱液,苯甲酸、山梨酸的回收率均接近于100%,当洗脱液达到4 BV时,3种添加剂均可以被完全洗脱,故先采用体积分数为60%的甲醇溶液洗脱,再用纯水清洗。

图3 洗脱液浓度对回收率的影响
2.3 检测条件优化
2.3.1 波长的选择 从图4~图6可以看出,苯甲酸的全波长扫描发现在对应的吸收波长为224 nm和283 nm有最大吸收峰。糖精钠也有两个较大的吸收峰,对应的吸收波长为272 nm和367 nm。山梨酸仅有一处最大吸收波长为254 nm。为兼顾3种食品添加剂的测定,选择230 nm作为检测波长。

图4 苯甲酸全波长扫描图

图5 糖精钠全波长扫描图
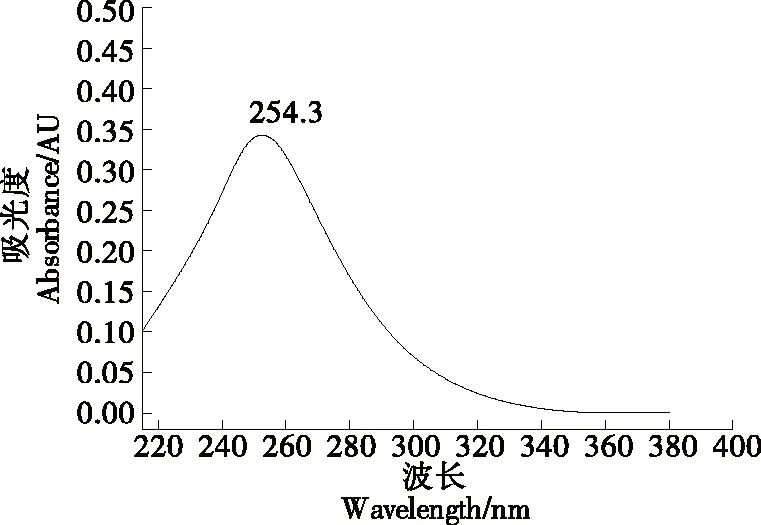
图6 山梨酸全波长扫描图
2.3.2 流动相优化 试验发现,当V乙酸铵∶V乙腈为75∶25时,各物质的分离效果最佳,各组分间分离度良好,峰型对称度良好(见图7),且各物质的保留时间最短。该检测条件下,苯甲酸的保留时间为2.409 min,山梨酸的保留时间为3.641 min,糖精钠的保留时间为6.507 min(见表1)。

图7 3种食品添加剂HPLC色谱图

表1 3种食品添加剂色谱数据表
2.4 方法学确认
2.4.1 线性范围与检测限 从表2可以看出,3种食品添加剂的质量浓度在0.5~10.0 mg/L时,所得到的线性回归方程具有良好的相关性,决定系数R2>0.990。在该检测范围内,对3种食品添加剂的检测限均达到了微克级别,均能够满足食品检测标准需要。3种食品添加剂的检测限均远低于产品控制的添加量限度75 mg/kg。其中,山梨酸检测限低于糖精钠和苯甲酸,为3.52×10-2mg/kg,低于GB 5009.28—2016执行的检出限0.005 g/kg,说明能得到更为精密的检测结果。

表2 3种食品添加剂线性回归方程和检测限
2.4.2 回收率与精密度 由表3可知,3种食品添加剂检测的平均回收率在95.44%~101.24%,相对标准偏差(RSD)在1.48%~2.92%,满足在RSD<5%的标准要求范围内。当质量浓度为0.05~0.50 mg/kg时,回收率为95.44%~99.53%,相对标准偏差≤2.46%;当质量浓度为0.50~1.00 mg/kg时,回收率为97.32%~101.24%,相对标准偏差≤2.92%。因此,该检测方法准确可靠,适用于同时分析卤制肉制品3种食品添加剂残留。
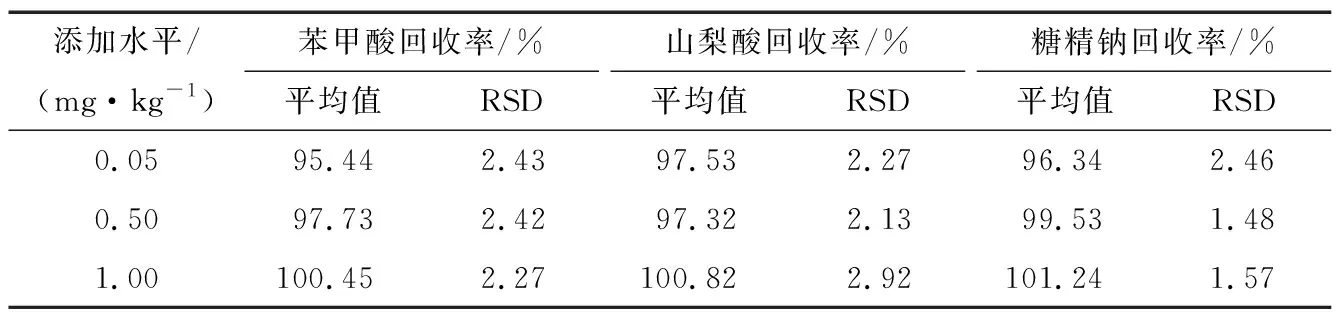
表3 3种添加剂在卤制肉制品中的添加回收率和相对标准偏差
2.4.3 实际样品定量分析 分别取3个不同厂家,不同批次的卤制肉样品共9份,按1.2.1样品前处理后,分别检测,检测结果见表4。

表4 实际样品检测结果
从购买样品的检测数据来看,酱卤肉中苯甲酸和山梨酸食品添加剂残留值均低于GB 5009.28—2016标准限值(0.075 g/kg),同时在个别包装中检出了糖精钠的残留,尽管残留值很低,仅有0.001 g/kg,但说明个别酱卤制肉生产厂家没有严格按照国家禁用要求进行管理。糖精钠曾被作为甜味剂,但因其安全性,中国于2015年禁止其在食品中使用[13]。糖精钠的检出,意味着某些食品厂家可能会利用仪器检测灵敏度的问题,在食品中添加一些国家违禁物来提升食品的风味。因此,建议相关部门建立严格的控制和检测标准,以保证食品安全。
3 结论
试验依据常规食品添加剂苯甲酸等物质结构特点选择特异性吸附的固相萃取剂SPE-MCX,有效防止了样品中脂肪的检测干扰,产品萃取回收率接近100%。采用HPLC来同时测定苯甲酸、山梨酸和糖精钠的残留,3种添加剂在质量浓度0.5~10.0 mg/L的范围内,其峰面积与质量浓度呈良好的线性关系,加标回收率为95.44%~101.24%,可以满足酱卤肉制品中多种添加剂快速分离检测要求。以该方法测定了不同批次的市售酱卤肉产品,发现现有的市售酱卤肉制品存在质量风险。后续将拓展样品的覆盖面,并对方法的使用稳定性和色谱柱的使用寿命等加以验证。
