超高效液相色谱—串联质谱法测定蜂蜜中10-羟基-2-癸烯酸
2021-09-21胡丽俐皮露露陈先桂
胡丽俐 黎 瑛 汪 辉 皮露露 陈先桂 蒋 莉
(长沙市食品药品检验所国家酒类产品质量监督检验中心,湖南 长沙 410006)
蜂蜜是一种营养丰富的天然食品,其中60%~80%是人体容易吸收的葡萄糖和果糖,还含有丰富的矿物质、维生素、氨基酸、酶等生物活性成分,具有抗菌消炎、抗氧化防衰老、抗癌增强免疫等生物功能,对骨质疏松症、胃肠道问题以及肝脏、心血管疾病有治疗作用[1]。随着消费者对蜂蜜需求量的增加,蜂蜜市场造假现象日趋严重,假蜂蜜不仅对人体的滋补效果较小,还容易引发肥胖、糖尿病、高血压、脂肪肝、急慢性肾损伤等疾病,严重危害消费者的身体健康[2]。同时,这种不正当的竞争损害了正规商户的利益,扰乱了正常的市场秩序。蜂蜜造假手段多样,常见的造假模式是以葡萄糖、果糖糖浆等为原料,添加色素、蜂蜜香精进行勾兑[3-4]。目前已建立了多种蜂蜜的鉴别检测方法,但大多数是基于蜂蜜外源性掺假物质成分的分析[5],而有关其内源性成分的分析方法较少。
10-羟基-2-癸烯酸(10-HDA)是由Butenandt从蜂王浆中分离出来,并且通过红外光谱确定了其结构[6]。10-HDA主要分泌于工蜂的上颚腺[7]。潘建国等[8-9]先后从蜂胶和蜂王胎中成功分离出10-HDA,平均含量分别为0.22%和0.15%。现有10-HDA的检测方法主要有高效液相色谱法[10-11]、气相色谱法[12]、气相色谱—质谱联用法[13]、薄层色谱法[14]、毛细管电泳法[15-16]、分光光度法[17]和红外光谱法[18]等。气相色谱法需进行甲基化衍生,操作繁琐;薄层色谱法易受试验环境等多因素影响,方法重现性差。以上方法仅适用于蜂王浆及其制剂中高含量10-HDA的测定,对于含量较低的蜂蜜难以达到检测要求。试验拟建立10-HDA的超高效液相色谱—串联质谱方法,并结合固相萃取技术实现蜂蜜中10-HDA的测定,以期为蜂蜜的真假鉴别提供一种内源性成分分析方法。
1 材料与方法
1.1 材料与试剂
10-羟基-2-癸烯酸(10-HDA)标准品:纯度>97.5%,中国食品药品鉴定研究院;
乙酸铵:分析纯,国药集团化学试剂有限公司;
甲醇、乙腈:色谱纯,德国Merck公司;
试验用水:超纯水,美国Millipore超纯水仪制备;
固相萃取柱:ProElut PLS 200 mg/6 mL,北京迪马科技有限公司;
蜂蜜:市售。
1.2 仪器与设备
超高效液相色谱仪:1290 Infinity Ⅱ型,美国安捷伦科技有限公司;
三重四级杆串联质谱仪:6470型,美国安捷伦科技有限公司;
电子天平:XS205DU型,梅特勒—托利多国际贸易(上海)有限公司;
超声波清洗仪:KQ5200DE型,昆山市超声仪器有限公司;
固相萃取装置:HSE-24B型,天津市恒奥科技发展有限公司;
氮吹仪:N-ECAP45型,美国Organomation 公司;
涡旋混合器:SK-1型,江苏省金坛市医疗仪器厂。
1.3 方法
1.3.1 标准溶液的配制
(1) 标准储备液:准确称取10-HDA标准品10.56 mg,用甲醇溶解并定容至100 mL,配制成100 μg/mL的标准储备液,于-20 ℃避光保存。
(2) 标准中间液:准确吸取标准储备液1.00 mL,用甲醇稀释并定容至100 mL,配制成1.00 μg/mL的标准中间液,于-20 ℃避光保存。
(3) 溶剂标准工作液配制:准确吸取一定体积的1.00 μg/mL的标准中间液至10 mL容量瓶中,甲醇定容,配制成质量浓度分别为0.05,0.10,0.20,0.40,0.60,0.80 μg/mL的标准工作液,现用现配。
(4) 基质匹配标准工作液配制:将1.3.2净化处理后的基质溶液氮气吹干,分别准确吸取1.00 mL相应浓度的标准溶液复溶,配制成质量浓度分别为0.05,0.10,0.20,0.40,0.60,0.80 μg/mL的基质匹配标准工作液,经0.22 μm有机滤膜过滤,现用现配。
1.3.2 样品前处理
(1)提取:准确称取1.00 g试样置于10 mL螺旋盖聚丙烯离心管中,加入8 mL甲醇—水溶液(V甲醇∶V水=4∶6),涡旋溶解,超声提取10 min,用甲醇—水溶液定容至10 mL,待净化。
(2) 净化:准确吸取5 mL提取液,加入到预先用5 mL甲醇和5 mL水平衡的ProElut PLS 200 mg/6 mL固相萃取柱中,用5 mL水进行淋洗,弃去所有流出液,抽干固相萃取柱,再用5 mL甲醇进行洗脱,收集洗脱液,于40 ℃水浴下氮气吹至近干,加入甲醇溶解并定容至1 mL,涡旋混匀1 min,经0.22 μm有机滤膜过滤,供超高效液相色谱—串联质谱测定。
1.3.3 前处理条件优化 按1.3.2的方法对样品前处理过程中的提取溶剂(V甲醇∶V水=2∶8,3∶7,4∶6,5∶5,6∶4,7∶3,8∶2)进行优化。
1.3.4 液相色谱条件优化 采用Agilent ZORBAX SB C18色谱柱(50 mm×2.1 mm, 1.8 μm)对目标物进行分离,优化有机相和水相溶液,通过比较峰形和响应,选择合适的流动相(乙腈—水、甲醇—水、乙腈-0.1%甲酸水、甲醇-0.1%甲酸水、乙腈-5 mmol/L乙酸铵、甲醇-5 mmol/L乙酸铵)。优化梯度洗脱程序,确定液相色谱条件。
1.3.5 质谱条件优化 分别在电喷雾正离子(ESI+)和电喷雾负离子(ESI-)模式下进行Scan扫描,得到对应的质谱总离子流图,确定化合物的母离子。对选定的母离子进行Product ion扫描,得到二级质谱图,选取丰度最强的碎片离子作为定量离子,次强的碎片离子作为定性离子,分别对子离子的碰撞能进行优化。在多反应监测模式(MRM)下,优化干燥气温度、干燥气流量、雾化气压力、鞘气温度、鞘气流量、毛细管电压、喷嘴电压等参数。
1.3.6 方法学验证
(1) 基质效应:分别用甲醇和1.3.2净化处理后的基质溶液制备相同浓度的低、中、高3个水平的标准工作溶液,评价基质效应。
(2) 线性范围、检出限和定量限:配制质量浓度分别为 0.05,0.10,0.20,0.40,0.60,0.80 μg/mL的基质匹配标准工作液,绘制基质匹配标准曲线。取代表样品,逐步定量地添加标准溶液,按1.3.2的方法进行处理,信噪比>3时的添加量为检出限,信噪比>10时的添加量为定量限。
(3) 准确度和精密度:取代表样品进行低、中、高3个浓度水平添加,每个添加水平平行测定6次,计算回收率。同时对等分样品进行6次平行测定,评估目标物的日内精密度。连续3 d内,对等分样品进行6次平行测定,评估目标物的日间精密度。
(4) 干扰试验:在代表样品提取液中加入喹诺酮类、四环素类、氯霉素类药物,配成最终质量浓度均为0.10 μg/mL的溶液,用试验方法进行分析,考察其他物质对目标物是否存在干扰。
1.3.7 数据处理 测量数据由Data Acquisition 进行采集,定量软件Quantitative Analysis 10.0 分析,使用Origin软件作图。
2 结果与分析
2.1 前处理条件的确定
试验发现,当样品提取液经过固相萃取小柱时,强极性物质如果糖和葡萄糖等直接流出,目标物10-HDA被吸附在小柱上,先用水淋洗将样品中残留的强极性杂质去除,再用甲醇将10-HDA 洗脱下来。当提取溶剂中甲醇体积分数≤40%时,提取液经ProElut PLS固相萃取小柱的流出液和水淋洗液中均未检测到10-HDA;当V甲醇∶V水=5∶5时,流出液中检出10-HDA。故采用甲醇—水溶液(V甲醇∶V水=4∶6)作为提取溶液。
2.2 液相色谱条件的确定
结果表明,采用乙腈—水为流动相体系时,10-HDA的响应值最高,但峰形不佳;采用甲醇—水为流动相体系时,10-HDA的响应一般,且峰形较差;采用乙腈-0.1%甲酸水和甲醇-0.1%甲酸水为流动相体系时,10-HDA离子化受到抑制,响应明显降低;采用乙腈-5 mmol/L乙酸铵、甲醇-5 mmol/L乙酸铵为流动相体系时,均能得到良好的峰形、最佳的分离效果和稳定的响应值(见图1)。由于甲醇毒性较小且成本较低,因此选择5 mmol/L乙酸铵(A)—甲醇(B)作为流动相,优化梯度洗脱程序见表1,色谱柱温度为35 ℃,进样量为5 μL。

1. 10-HDA a. 乙腈-0.1%甲酸 b. 乙腈-5 mmol/L乙酸铵 c. 甲醇-5 mmol/L乙酸铵 d. 乙腈-水 e. 甲醇-0.1%甲酸 f. 甲醇—水

表1 梯度洗脱程序
2.3 质谱条件的确定
10-HDA的分子式为C10H18O3,相对分子质量为186.2。试验过程中, ESI+、ESI-模式下准分子离子峰[M+H]+和[M-H]-的m/z分别为187.2,184.9,ESI-模式下的响应值高于ESI+的,故采用ESI-模式。在ESI-模式下,选择m/z为184.9的离子作为母离子,进行Product ion扫描,得到10-HDA的二级质谱图。10-HDA在碰撞室中主要碎裂成m/z为138.8,110.8的碎片,推测其可能的裂解途径为图2。选择184.9/138.8、184.9/110.8离子对作为多反应监测(MRM)的离子对,其中184.9/138.8的响应值更高,故将其作为定量离子对,184.9/110.8作为定性离子对。在多反应监测(MRM)下优化各项参数,得干燥气温度为325 ℃、干燥气流量为8 L/min、雾化气压力为0.31 MPa、鞘气温度为400 ℃、鞘气流量为11 L/min、毛细管电压为-3 500 V、喷嘴电压为500 V,目标物的相关质谱参数见表2。

图2 10-HDA可能的裂解途径

表2 10-HDA的质谱参数
2.4 方法学验证
2.4.1 基质效应评价 样品基质中的干扰物质对目标物电离效率的影响用基质效应(ME)来评价。当|ME|≤20%时,基质效应对结果的准确性影响不大,可以忽略不计;当|ME|>20%时,基质效应较强,对定量结果会产生较大干扰。由表3可知,蜂蜜中10-HDA在低、中、高3个浓度水平的ME为-29.9%~-45.5%,存在明显基质抑制效应。因此,采用基质匹配标准曲线来定量,以获得更加准确的结果。
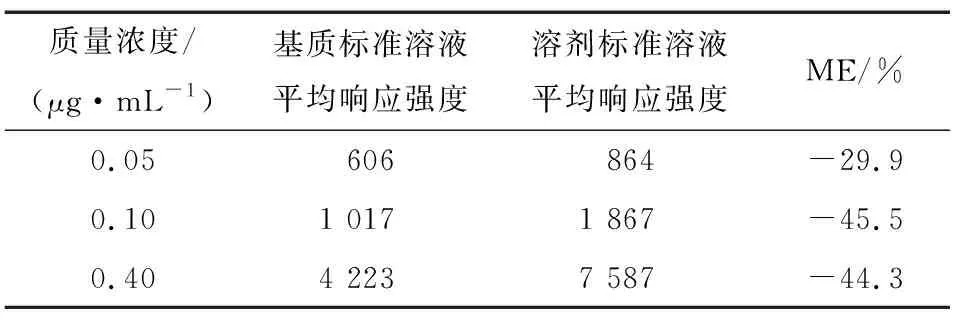
表3 蜂蜜中10-HDA的基质效应
2.4.2 线性范围、检出限和定量限 10-HDA的基质匹配标准溶液在0.05~0.80 μg/mL质量浓度范围内呈良好的线性关系,线性回归方程为Y=12.4X-283.7,相关系数为0.995。方法的检出限为 0.03 mg/kg,定量限为 0.10 mg/kg。
2.4.3 准确度和精密度 在0.10,0.20,1.00 mg/kg添加浓度水平下,10-HDA的平均回收率为96.9%~108.3%,相对标准偏差为4.5%~10.7%。由表4可知,日内精密度和日间精密度分别为5.9%~9.2%和3.5%~14.3%。

表4 方法的回收率与精密度
2.4.4 干扰试验 文献[19-20]报道,蜂蜜中可能有喹诺酮类、四环素类、氯霉素类、硝基呋喃类药物残留。采用试验方法分析含有以上药物的溶液,在目标物保留时间区域未发现干扰,将以上药物加入到代表性样品中,目标物浓度未发生改变[21]。因此,试验方法有良好的抗干扰能力。
2.5 实际样品分析
应用试验方法对12个蜂蜜样品进行检测,由图3可知,除1个假蜂蜜样品未检出10-HDA外,其余11个蜂蜜样品中均有检出10-HDA,最高含量为0.96 mg/kg,最低含量为0.22 mg/kg,说明10-HDA作为内源性成分在蜂蜜中普遍存在的。

图3 基质标准溶液和典型样品溶液MRM色谱图
3 结论
建立了蜂蜜中10-羟基-2-癸烯酸的超高效液相色谱—串联质谱检测方法。该方法操作简便,无需进行气相色谱法繁琐的衍生化处理;灵敏度高,检出限和线性范围等参数均优于高效液相色谱法;且重现性好,抗干扰能力强于薄层色谱法。试验方法能够满足蜂蜜中10-羟基-2-癸烯酸的定性定量分析。由于蜂蜜中10-羟基-2-癸烯酸的含量受蜜源种类、地理环境等因素影响,后续将加大对不同种类和产地的蜂蜜监测,完善蜂蜜中10-羟基-2-癸烯酸含量的数据。
