Cr和C共掺TiO2的电子结构和光学性质的研究
2021-09-16朱海霞
周 悦, 吴 洁, 朱海霞
(盐城师范学院 物理与电子工程学院, 盐城 224051)
1 引 言
在金属氧化物半导体家族中,二氧化钛(TiO2)由于其化学热稳定性高、催化活性强、无毒等优点,已经被最广泛使用,如利用光照可以降解环境污染物、分解和净化水、实现光电转换等等. 然而,锐钛矿型TiO2的本征带隙为~3.2 eV,这意味着锐钛矿型TiO2只能被太阳光的紫外线激发,为了提高TiO2光电能量转换性能,如何调整TiO2的带隙值达到其理想状态下的~2.0 eV,实现可见光激发吸收,是许多实验和理论工作者感兴趣的问题. 而掺杂是一种最简单最有效的调节TiO2的带边和带隙值的方法之一[1-3]. 由于TiO2的价带顶主要是由O的p轨道构成,导带低主要是由Ti的d轨道构成,所以掺杂通常会选择那些p轨道能高于O-2p轨道能或d轨道低于Ti-3d轨道能,如N、S、B和C等非金属元和Mo,W,V和Cr过渡金属元素素[4-6]. 如C掺杂TiO2可显著改善TiO2的可见光吸收[7, 8]. 前人实验合成了C掺杂的TiO2,发现它促进了可见光的光催化活性,并在可见光区域显示出两个光学吸收峰[9]. 据报道,过渡金属元素Cr掺杂的TiO2也可以有效地增强可见光光催化活性,因为Cr-3d电子引入的局部间隙状态降低了TiO2的带隙[10]. 也有人使用组合溶胶-凝胶和水热法,合成测量Cr掺杂锐钛矿TiO2的体系,其中Cr原子取代Ti原子,增强了可见光吸收以及可见光光催化活性[11]. 然而非金属单掺会存在电子部分占据局域间隙态这一严重问题[7],即这些带隙态易成新增的电子-空穴复合中心,直接减缓减少光生电子向TiO2表面迁移及参与光催化反应,从而降低TiO2光催化量子效率. 为了克制这种复合中心,就出现了共掺杂:如W和N共掺TiO2,其带隙值有效减小,提高光生电子对的有效分离. 有工作用溶胶-凝胶法合成Cr-N共掺杂TiO2,其样品具有很强的太阳光吸收激发. 还有人对Nb-C、Mo-C、Fe-N、Mo-N共掺杂TiO2进行了第一性理论研究,发现共掺TiO2体系将更有利于出现可见光光催化[6, 12-13].
根据上述分析,可以预期 Cr和C共掺TiO2可能会导致很好的可见激发吸收和催化活性. 尽管已有一些涉及Cr-C共掺杂TiO2的研究工作,但是到目前为止,对Cr-C不同掺杂TiO2构型的电子结构和光学性质的全面系统理论研究仍然缺乏. 在本论文中,我们选择锐钛矿型TiO2为研究对象,主要基于密度泛函理论的第一性计算来研究Cr和C共掺杂锐钛矿TiO2的电子结构与光学特性. 文中将主要讨论两种非等价的Cr和C相邻共掺杂TiO2的构型. 具体详细讨论了带隙变窄的微观机制和增强的可见光光催化活性的微观机制.
2 理论模型构建和计算细节
2.1 模型构建
我们首先将锐钛矿型TiO2的原胞扩展成2×2×1含有48个原子的TiO2,选用超胞来模拟Cr和C共掺杂构型,这也是常用的模拟掺杂超胞的尺寸[6],对于Cr和C共掺情况,我们发现Cr和C非近邻掺杂TiO2的总能量都远远大于Cr和C近邻共掺的. 因此Cr和C近邻补偿共掺TiO2体系是最稳定,所有我们主要讨论Cr和C紧邻结合的近邻补偿共掺情况,通常这种结合也具有最低总能量[6,12]. 于是我们的掺杂模型是这样构建的:我们首先用一个Cr原子去代替2×2×1锐钛矿型TiO2超晶格中的一个Ti原子,此时Cr原子是位于一个氧八面体的中心,相对于Cr原子而言,氧八面体的赤平面上有四个氧离子位置,其两顶角位置个一个原子. 因此,C我们选择两种O位替代,一种选择氧八面体的赤平面上的一个O位替代,另一种选择氧八面体顶点上的一个氧位替代. 这样我们就可以得到两种类型的的Cr-C近邻补偿共掺杂锐钛矿TiO2构型,其构型图如图1(a~b)所示. 这两种Cr-C紧邻补偿共掺杂TiO2构型分别表示为:CrC1-TiO2、CrC2-TiO2.

图1 2×2×1 掺杂锐钛矿型TiO2 超胞结构图(a) CrC1-TiO2, (b)CrC2-TiO2 Fig. 1 Structure schemas of the 2×2×1 doped TiO2 supercell (a) CrC1-TiO2, (b)CrC2-TiO2
2.2 计算方法和计算参数设定
本文所有计算都是基于密度泛函理论框架下第一性原理计算所得,第一性原理计算软件选用的是商业计算模拟软件包Vienna ab initio (vasp5.12)[14,15]. 利用投影缀加波赝势处理离子核与价电子之间的互作用[16]. 交换相关势采用的是广义梯度近似GGA(Generalized Gradient Approximation)的PBE (Perdew-Becke-Erzenhof)形式的泛函[17]. 对于晶体的布里渊区倒格空间的积分,我们使用的是Monkhorst-Pack网格方案[18],在文中,对于我们所讨论锐钛矿型2×2×1的TiO2超胞,其第一布里渊区内k点取为5×5×5的Monkhorst-Pack网格[18]. 计算中设置能量收敛阈值为1.0×10-6eV/atom. 在优化结构时,所以掺杂的TiO2体系的晶格参数合每个原子的位置都全面放开进行优化. 直到作用在超胞上每个原子上的Hellmann-Feynman力降低到1.0 meV/Å才停止优化.
3 结果与讨论
3.1 电子结构
首先我们计算了纯TiO2, CrC1-TiO2和CrC2-TiO2.体系的能带结构图如图2所示,图中红色虚线为费米能级位置. 对于纯TiO2体系,是一种直接带隙的半导体,其最高的O-2p带与最低的Ti-3d带之间的带隙为Eg=2.03 eV. 此结果与以前的用GGA方法所得到的理论结果相一致[6]. 由于GGA方法的本身的局限性,致使理论结果与TiO2实验带隙值(3.2 eV)相比是低估了带隙值. 图3(b)是CrC1-TiO2构型的能带结构图,其费米能量位于价带顶上方,显示出了半导体特性. 这共掺杂TiO2中的价带顶主要由C-2p轨道态及与其紧邻Cr-3d轨道的耦合态. 由于姜-泰勒变形,Cr-3d轨道的t2g(dxy,dxz,dyz)轨道进一步分裂成Cr-dxy,Cr-dxz,Cr-dyz态. 导带底(CBM)附加带主要由Cr-3dyz轨道态构成. 我们可以看到CrC1-TiO2构型有效带隙为1.19 eV. CrC1-TiO2构型带隙与纯TiO2相比减小了0.84 eV. 可以看出CrC1-TiO2构型将会显著增TiO2体系的可见光激发吸收活性. 图3(c)是CrC2-TiO2.的带结构,其费米能量Ef仍然是位于价带顶上方,但带结构特征与CrC1-TiO2构型显著不同. 除纯TiO2带边外,带隙中有深带隙态存在,带隙态主要由C-2p轨道和Cr-3d轨道耦合而成. 这主要由于CrC2-TiO2.构型较CrC1-TiO2构型的氧八面体的晶体场扭曲的更严重,所以CrC2-TiO2.构型具有更强的姜-泰勒变形效应,Cr-3d进一步降低简并态从而能级进一步分裂,其深间隙状态主要由Cr-3d轨道分裂成的Cr-3dxy和Cr-3dyz贡献. 由于深间隙态的存在,电子从价带顶转移到导带底的所需要的能量就大大减小. 从图中可以看到,价带顶到深带隙的能量宽度为0.84 eV,再从深带隙到导带底的能量宽度为0.51 eV. 这都将有效提高TiO2的可见光吸收活性.

图2 TiO2的不同掺杂体系的能带结构 (a)pureTiO2 , (b) CrC1-TiO2 , (c)CrC2-TiO2. 红色虚线为费米能级位置. Fig. 2 Band structures of TiO2 supercell with different doping methods (a)pure TiO2 , (b) CrC1-TiO2 , (c)CrC2-TiO2.
为了进一步研究Cr与C掺杂TiO2体系的性质,我们计算了纯TiO2, CrC1-TiO2和CrC2-TiO2的总态密度(TDOS)和分态密度(PDOS). 其分布图如图3所示,各分图中的黑色点划线是纯TiO2系统对应的TDOS分布,绿色实现对于的是掺杂后TiO2体系的TDOS分布. 图中绿色的垂直虚线代表电子的最高占据能级位置. 从图中可以看出,掺杂后的体系几乎没有改变纯TiO2体系所固有的带隙值. 改变Cr与C掺杂体系的有效带隙值得主要原因是掺杂原子所引入的带隙子带,首先看图4(a),在价带上方存在 三条主要由C-2p轨道和Cr-3d轨道耦合而成的杂化轨道. 由于姜-泰勒变形,Cr-3d轨道的t2g轨道进一步分裂的Cr-3dyz轨道态在导带底下方又形成一附加子带. 对于CrC2-TiO2构型,在价带上方也存在主要由C-2p轨道和Cr-3d耦合的子带,导带底下方形成一主要由Cr-3d轨道进一步分裂的Cr-3dyz而产生的附加子带. 此外体系存在一个深带隙态,主要由Cr-3dxz贡献产生. 可以看出,正是由于这些掺杂引入了各种带隙态,将有效减小电子从价带顶想导带跃迁的所需能量,将有效提高体系的可见光的光电转换效率.

图3 TiO2的各种掺杂体系的TDOS和PDOS图 (a) CrC1-TiO2, (b) CrC2-TiO2, 图中红点划线代电子的最高占据能级位置Fig. 3 TDOS and PDOS plots of TiO2 with different doping methods (a) CrC1-TiO2, (b) CrC2-TiO2
3.2 光学特性
为了进一步分析Cr和C掺杂TiO2体系光学性质,我们计算了纯TiO2, CrC1-TiO2和CrC2-TiO2的复介电函数和吸收系数.
图4是计算得到的CrC1-TiO2和CrC2-TiO2体系的复介电函数分布图,从图中看出Cr与C共掺杂体系得到的介电性能随光子能量变化大趋势大体是比较类似的. 其实部和虚部主要的介电峰都存在三个. 对于CrC1-TiO2体系,介电函数的实部最大介电峰出现在2.34 eV附近,峰值约为9.13,其他介电峰位置不太明显. 虚部在1.4 eV处有个小介电峰,其峰值约为0.4,虚部最高的第一介电峰位于3.82 eV,其峰值约为9.65, 实部在4.87 eV附近降低为0,虚部在10.1 eV附近降低为0. CrC2-TiO2体系,其介电函数的实部最大介电峰出现在2.92 eV附近,峰值约为9.23. 虚部最高的介电峰位于3.68 eV,其峰值约为9.94, 实部在4.82 eV附近降低为0,虚部在10.2 eV附近降低为0. 在0-2.5 eV能量范围内,CrC1-TiO2和CrC2-TiO2体系的介电函数虚部都远远大于纯TiO2体系. 说明掺杂后TiO2体系将提高了对光的利用率.
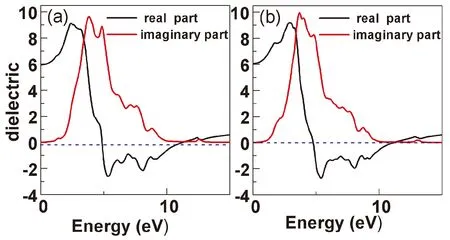
图4 TiO2的不同体系的介电函数图(a) CrC1-TiO2, (b)CrC2-TiO2Fig. 4 The dielectric functions of (a) CrC1-TiO2, (b)CrC2-TiO2
我们计算所得纯TiO2, CrC1-TiO2和CrC2-TiO2体系的光吸收谱曲线分布如图5所示,太阳光中可见光的波长分布在390 nm~760 nm区域,即光量子的能量在1.64 eV~3.19 eV范围之内,其内插图是把声子能量设定在可见光范围的光吸收谱. 纯TiO2吸收光谱为图中红色点划线. 我们现纯TiO2体系的光吸系数在可见光范围内非常小,几乎没什么可见光激发活性,只有紫外激发活性. 比较发现CrC1-TiO2和CrC2-TiO2体系光谱吸收边,会发现其吸收边都沿展到了可见光区域内,并形成了较宽的可见光吸收区域,也就是说,Cr与C共掺杂TiO2实现了预期的光吸收边红移,比较发现CrC1-TiO2和CrC2-TiO2体系,我们发现CrC2-TiO2的光吸收系数 (图中红色实线) 整体上比CrC1-TiO2的吸收系数(图中蓝色实线)要大,特别是在可见光吸收范围内,这表明,对于Cr与C共掺杂TiO2体系,其CrC2-TiO2掺杂构型更加有利于提高TiO2体系的可见光催化活性,这可能是电子结构上的细微差异导致的. 总之,通过光吸收谱的计算和分析,进一步证实Cr与C共掺杂TiO2是一种有效提高TiO2体系可见光光催化活性的方法,增强了TiO2的可见光光电转换效率.

图5 纯TiO2, CrC1-TiO2和CrC2-TiO2的光吸收谱. Fig. 5 The optical absorption spectra of pure TiO2, CrC1-TiO2 and CrC2-TiO2
4 结 论
利用第一性原理计算,研究了Cr与C共掺锐钛矿型TiO2的的电子结构和光学性质,能带的结构,态密度和光学性质. 主要讨论了两种种可能的Cr和C相邻共掺杂TiO2构型CrC1-TiO2和CrC2-TiO2. CrC1-TiO2体系在价带上方出现了主要由C-2p轨道和Cr-3d轨道耦合成的子带. 有效带隙较纯TiO2相比变窄了0.84 eV. 同时,由于掺杂原子C在一定度上破坏纯TiO2所具有的氧八面体的晶体场,产生Jahn-Teller效应,Cr-3d轨道的t2g轨道进一步分裂的成Cr-3dyz轨道在导带底形成附加带. 对于CrC2-TiO2体系,其带隙中有深带隙态存在,由于深间隙态的存在,价带顶到深带隙的能量宽度为0.84 eV,电子从价带顶转移到导带底的所需要的能量将大大减小. 此外,沿着掺杂离子Cr-C,存在一定的极化,这可以在CrC-TiO2系统内部形成局域的极化场这种内部极化场将有利于光生电子与空穴的分离,最后,我们的计算光吸收谱显示Cr与C共掺TiO2的光学边延伸到了可见关区域,增强了可见光活性,大大提高了太阳光的利用率.
