本征及掺杂石墨烯对2,3,7,8-四氯二苯并呋喃吸附机理的计算模拟研究
2021-09-16宋小东林小龙曾巧玲孙玉希
王 群 , 宋小东 , 林小龙 , 刘 佳 , 曾巧玲 , 孙玉希
(1. 绵阳师范学院 生命科学与技术学院, 绵阳 621006 ; 2. 绵阳师范学院 光致功能材料重点实验室, 绵阳 621006;3. 山东省曲阜师范大学 生命有机分析重点实验室, 曲阜 273165; 4. 北京市食品风味化学重点实验室, 北京 100048)
1 引 言
二噁英是一类剧毒有机污染物, 它是多氯代二苯并-对-二噁英 (PCDDs)和多氯代二苯并呋喃(PCDFs)的统称. 二噁英物质可以使人致癌, 引发严重的发育问题, 损害免疫系统, 并干扰荷尔蒙系统, 严重威胁着人类的健康[1, 2]. 在二噁英家族中, 2,3,7,8 -四氯二苯并呋喃(2,3,7,8-TCDF)是最常见的有毒物质之一, 由两个苯环和一个呋喃氧原子组成, 苯环周围有四个氯原子. 在生活垃圾, 工业垃圾及医疗废弃物的焚烧过程中均可形成TCDF二噁英污染物[3, 4]. 由于二噁英对人体和环境具有很高的毒性, 因此开发一种简单有效的方法来检测和去除二噁英是相当有必要的. 近年来, 对二噁英类有毒物质处理的方法主要有吸附法[5], 光降解法[6], 生物分解法[7]及化学分解法[8]等. 其中吸附法以效率高, 工艺简单, 操作方便, 成本低等优点受到了广大研究者的关注. 目前吸附材料主要有新型的碳纳米材料, 比如: 碳纳米管[9], 石墨烯[10]等.
石墨烯是sp2杂化的一种二维碳纳米材料, 由于其良好的电学, 光学, 磁学特性及巨大的比表面积, 在气体传感器, 电子器件, 药物运输等领域均有广泛的应用前景[11]. 大量研究表明石墨烯可被用于多种分子的高灵敏传感器[12, 13]. 然而, 文献表明, 吸附剂与石墨烯基底及其表面主要是非共价相互作用, 作用力较弱, 因而限制了它在传感器中的应用[14]. 要解决此问题, 研究者们多采用对石墨烯掺杂改性的方法提高其反应活性. Hong等人[2]研究了石墨烯和碳纳米管对2,3,7,8-四氯二苯并-对-二噁英( TCDD)的吸附, 发现Ca掺杂石墨烯可以增加TCDD在石墨烯和碳纳米管之间的相互作用, 这是因为它们之间形成了π-Ca-π相互作用. Zhang等人[13]采用密度泛函理论理论探讨了Ti, N和Ag原子掺杂石墨烯对2,3,7,8-TCDD的吸附, 发现Ti掺杂的石墨烯有最强的吸附能力. Zhou等人[15]研究了Mn和Fe掺杂石墨烯对2,3,7,8-TCDF的吸附, 发现掺杂大大提高了对剧毒有机分子的吸附.
就我们所知, 2,3,7,8-TCDF这种二噁英污染物在掺杂石墨烯表面的吸附情况如何?详细的反应机理如何?这些问题目前还鲜有报道. 而且由于二噁英类物质的剧毒性, 限制了它们在实验方面的研究, 因此采用计算模拟的方法不失为一种有效的方法. 在本项研究中, 我们探索了本征石墨烯与Ti, Fe及Pt原子掺杂石墨烯对有毒气体2,3,7,8-TCDF的吸附, 从吸附能, 电荷转移, 态密度及差分电荷密度等方面探讨了本征及掺杂石墨烯对2,3,7,8-TCDF二噁英吸附的可能性, 希冀为有害气体二噁英的检测及去除提供有效的参考.
2 模拟细节
2.1 模型的建立
2,3,7,8-TCDF模型: 分子式为C12H4OCl4, 含有两个苯环, 一个呋喃基的氧原子, 四个氯原子, 结构优化之后如图1所示.

图 1 结构优化之后2,3,7,8-TCDF二噁英的三维结构. (碳原子: 黑色; 氧原子: 红色; 氢原子: 灰色; 氯原子: 绿色).Fig. 1 Three-dimensional structures of 2,3,7,8-TCDF after geometric structure optimization. (Carbon: black; Oxygen: red; Hydrogen: gray; Chlorine: green).
本征石墨烯, 金属原子掺杂石墨烯模型: 本征石墨烯模型是通过石墨切面获得, 首先构建一个石墨烯晶胞, 赋予石墨烯周期性, 并于其上方加上厚度为30 Å的真空层, 优化之后的C-C键长大约为1.420 Å[16], 如图2a所示. Ti, Fe及Pt金属原子掺杂石墨烯是在本征石墨烯中将一个碳原子替换成一个金属原子而获得, 结构优化之后如图2b-2d所示.
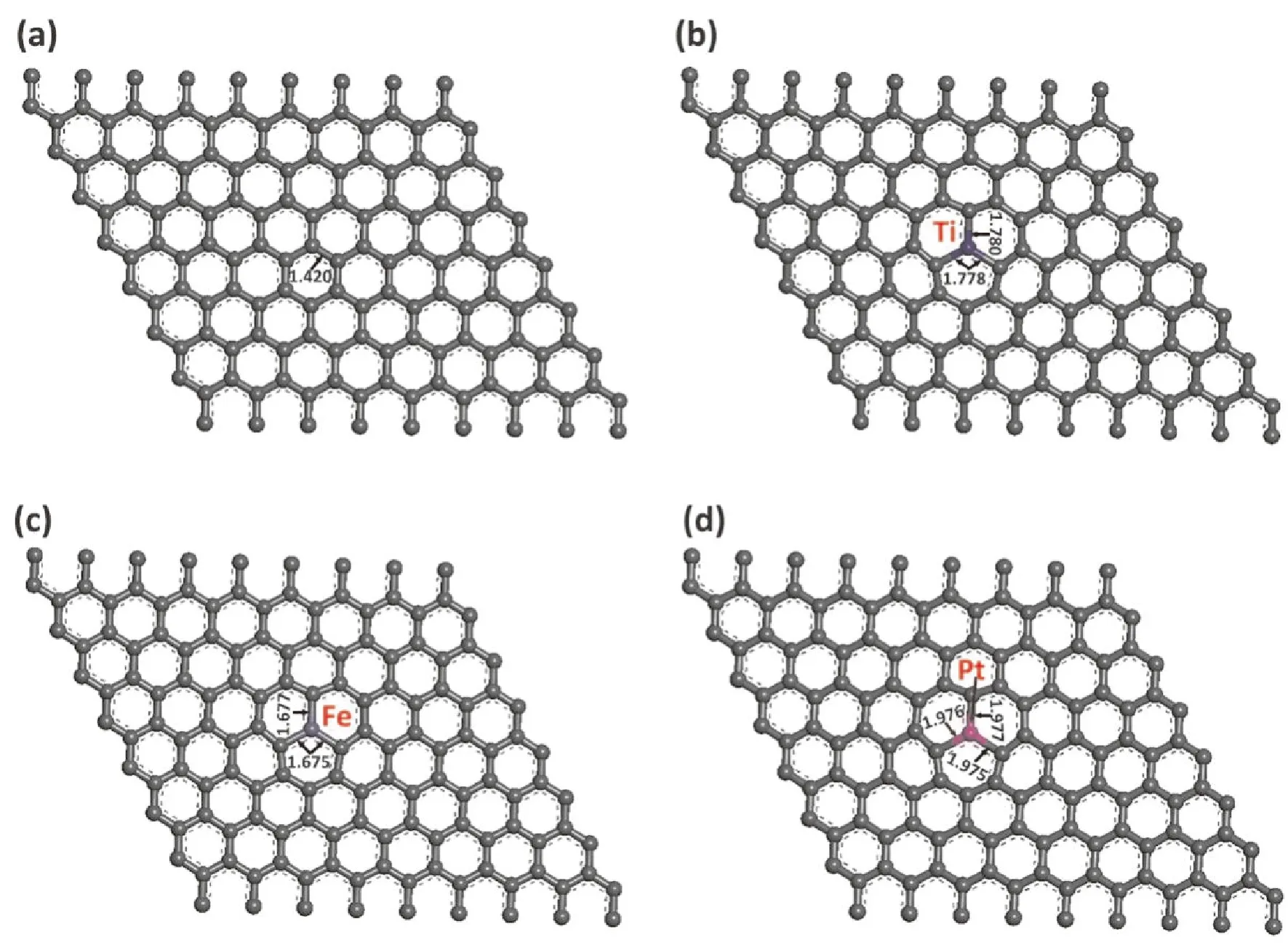
图 2 结构优化之后的三维结构. (a) 本征石墨烯; (b) Ti掺杂石墨烯; (c) Fe掺杂石墨烯; (d) Pt掺杂石墨烯. (钛原子: 蓝色; 铁原子: 浅灰色; 铂原子: 粉色). 键长单位为Å.Fig. 2 Three-dimensional structures of (a) intrinsic graphene; (b) Ti-doped graphene; (c) Fe-doped graphene; (d) Pt-doped graphene after geometric structure optimization. (Titanium (Ti): blue; Iron (Fe): light gray; Platinum (Pt): pink). Bond lengths are in Å.
TCDF-G相互作用模型: 根据文献, 发现TCDF平躺于各种石墨烯表面有最大相互作用的可能性[15], 因此, 我们建立了TCDF平躺于四种石墨烯表面的模型, 结构优化之前如图3所示, 优化之后如图4所示.
图 3 结构优化之前, TCDF与 (a) 本征石墨烯; (b) Ti掺杂石墨烯; (c) Fe掺杂石墨烯; (d) Pt掺杂石墨烯相互作用.Fig. 3 Before the optimization, TCDF interacted with the (a) intrinsic graphene; (b) Ti-doped graphene; (c) Fe-doped graphene; (d) Pt-doped graphene.
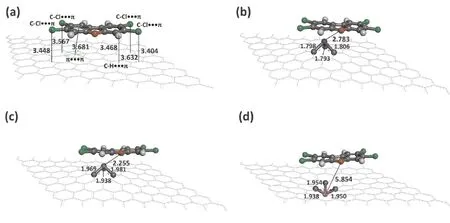
图 4 结构优化之后, TCDF与 (a) 本征石墨烯; (b) Ti掺杂石墨烯; (c) Fe掺杂石墨烯; (d) Pt掺杂石墨烯相互作用. 键长单位为Å.Fig. 4 After the optimization, TCDF interacted with the (a) intrinsic graphene; (b) Ti-doped graphene; (c) Fe-doped graphene; (d) Pt-doped graphene. Bond lengths are in Å.
2.2 模型参数
本论文所有的计算均使用Materials Studio (Accelrys, San Diego, CA)中基于第一性原理DFT的Dmol3软件进行研究. 我们采用Perdew-Burke-Ernzerhof (PBE)广义梯度近似 (GGA)计算交换关联能[17]. Quijano-Briones等人[18]报道了PBE函数中色散力校正(dispersion-corrected density functional theory, DFT-D)被认为是最适用于苯环类分子与石墨烯相互作用的理论研究. 因此, 此研究中所有结果都是使用DFT-D校正得到. 我们采用等同于Gaussian中6-31G**基组的DNP双重数值基组 (DNP double numerical basis set)进行计算, 此基组不仅计算成本低, 而且计算结果准确度高[19, 20]. 我们采用DFT半核赝势 (semicore pseudopotentials) 处理内层电子. 对于布里渊区域 (Brillouin zone)内的K点采样, 我们的Monkhorst-Pack方案中网格设置为6×6×1[21], 费米拖尾效应 (Fermi smearing)值设置为0.005 Ha (1Ha = 27.2114 eV), 全局轨道截断 (global orbital cutoff)值设置为5.0 Å. 所有的原子是被允许放松的. 体系的几何构型优化和能量计算收敛条件为: (a)自洽循环数量级≤1.0×10-6Ha/atom;(b)能量值数量级≤1.0×10-5Ha /atom;(c)最大应力≤0.002 Ha /Å;(d)最大位移≤0.005 Å. 我们根据公式(1)分析了TCDF吸附在石墨烯表面的吸附能Eads:
Eads=ETCDF+surface-(ETCDF+Esurface)
(1)
此时ETCDF+surface代表TCDF与各种石墨烯表面的吸附能,ETCDF代表单个TCDF的吸附能,Esurface代表单个石墨烯表面的吸附能. 本研究中吸附能的值如表1所示, 吸附能越负, 则表示吸附体系越稳定[22, 23].
3 结果与讨论
3.1 金属原子掺杂石墨烯结构的稳定性分析
结构优化之后, 金属原子掺杂的石墨烯如图2b-2d所示. 掺杂的金属原子没有突出石墨烯平面, C原子与金属原子之间的距离均有所增加. 其中, Ti与邻近的三个C原子之间距离分别为1.778, 1.778和1.780 Å, Fe与邻近的三个C原子之间距离分别为1.675, 1.675和1.677 Å, Pt与邻近的三个C原子之间距离分别为1.975, 1.976及1.977 Å, 这些键长与文献值完美吻合[24-26]. 此外, 键长结果也表明金属原子与C原子之间的键未被破坏, 因此, 本研究中金属原子掺杂的石墨烯模型都是稳定的.
3.2 吸附能分析
各种体系的吸附能值如表1所示, 吸附能值越负表示这些吸附体系热力学上越稳定[22]. 当TCDF吸附于本征石墨烯表面时(图4a) , 吸附能为-0.98 eV, 表明TCDF可以稳定吸附其表面, 但是它们之间的相互作用较弱. TCDF中苯环与石墨烯苯环的最短距离为3.681 Å, TCDF中H原子离石墨烯平面的最短距离为3.468 Å, 四个氯原子离石墨烯平面的最短距离分别为3.404, 3.448, 3.567和3.632 Å, 如图4a所示. 这些距离值和吸附能值均表明TCDF和本征石墨烯之间形成了π…π, C-H…π和C-Cl…π非共价相互作用, 这也是和以前的报道一致的. Zhou等人[15]表明TCDF与本征石墨烯之间形成非共价相互作用, 最短的吸附距离为3.800 Å, 且它们之间的吸附能为-0.103 eV. Zhang等人[13]报道二噁英TCDD与本征石墨烯之间也是非共价相互作用, 最大的吸附能为-0.40 eV.
TCDF和Ti, Fe及Pt掺杂石墨烯表面相互作用如图4b-4d所示, 我们发现三种掺杂金属原子都突出于石墨烯表面, 从而破坏了石墨烯六方结构, 掺杂位点的活性发生了改变[16]. Ti及Fe掺杂石墨烯与TCDF之间的吸附能分别为-2.16和-2.58 eV, 表明Ti和Fe掺杂可以增加它们之间的相互作用, 此结论也是与文献一致的. Zhou等人[15]表明缺陷石墨烯掺杂Fe原子可以增加TCDF的吸附, Zhang等人[13]发现Ti掺杂石墨烯可以大大增加与TCDD的相互作用. 同时, 通过图4b和4c发现, 在相互作用之后, Ti-C和Fe-C之间的距离有所增加, 分别为1.793-1.806 Å (未相互作用之前为1.778-1.780 Å)及1.938-1.981 Å (未相互作用之前为1.675-1.677 Å) . Ti原子和Fe原子朝着TCDF方向移动, 与TCDF的呋喃氧原子之间形成Ti-O和Fe-O相互作用, 它们之间的相互作用距离分别为2.783和2.255 Å, 说明它们之间形成了典型的“metal-O”共价相互作用. 这种相互作用在提高掺杂石墨烯和有机分子相互作用的吸附中也会经常被报道. Wang等人[16]报道多巴胺和Fe, Ca掺杂石墨烯因存在着这种“metal-O”共价相互作用而使得多巴胺和掺杂石墨烯相互作用大大增强, 他们发现丝氨酸与Fe, Cr, Al, Mn和Ti原子掺杂石墨烯之间也出现了这种相互作用[26].
TCDF和Pt掺杂石墨烯之间的吸附能为-0.85 eV, 小于TCDF与本征石墨烯之间的相互作用. 图4d表明Pt原子掺杂的石墨烯表面向TCDF相反的方向被排斥到了平面之外. Pt-O之间的相互作用距离增大为5.854 Å, 因此它们之间的相互作用较弱. 这种现象也在文献中曾被发现. 王等人[27]发现Hg /Pd掺杂的石墨烯表面向苄硫醇相反的方向被排斥到了平面之外, 导致它们之间相互作用较小.
3.2 态密度的分析
基于能带理论的态密度(Density of states, DOS)表示某个能量状态下电子态的数目. 如果相邻原子的局域态密度在同一个能量上同时出现了尖峰, 则将其称之为杂化峰( hybridized peak), 我们可以较直观地判断相邻原子之间的作用强弱[27]. 在接下来的分析中, 主要分析了掺杂金属原子和呋喃氧原子之间相互作用情况, 如图5所示. 在TCDF-Ti-G模型中(图5a), Ti和O原子在-10到-2 eV之间有重叠杂化峰, 表明它们在此能量区域有相互作用的可能性. 在TCDF-Fe-G模型中(图5b), Fe和O原子在-10到+2 eV之间有重叠杂化峰, 表明它们在此能量区域有相互作用的可能性. 而在TCDF-Pt-G模型中(图5c), 在-10到+2 eV之间Pt和O原子重叠杂化峰较少, 说明它们之间存在相互作用的可能性较少. 同时, 我们发现在TCDF-Fe-G模型中(图5b), Fe和O原子杂化峰尖锐度变小, 表明它们的电子离域性变强, Fe原子与呋喃O原子相互作用可能性也更大[28]. 态密度的结果与吸附能的结果相一致.
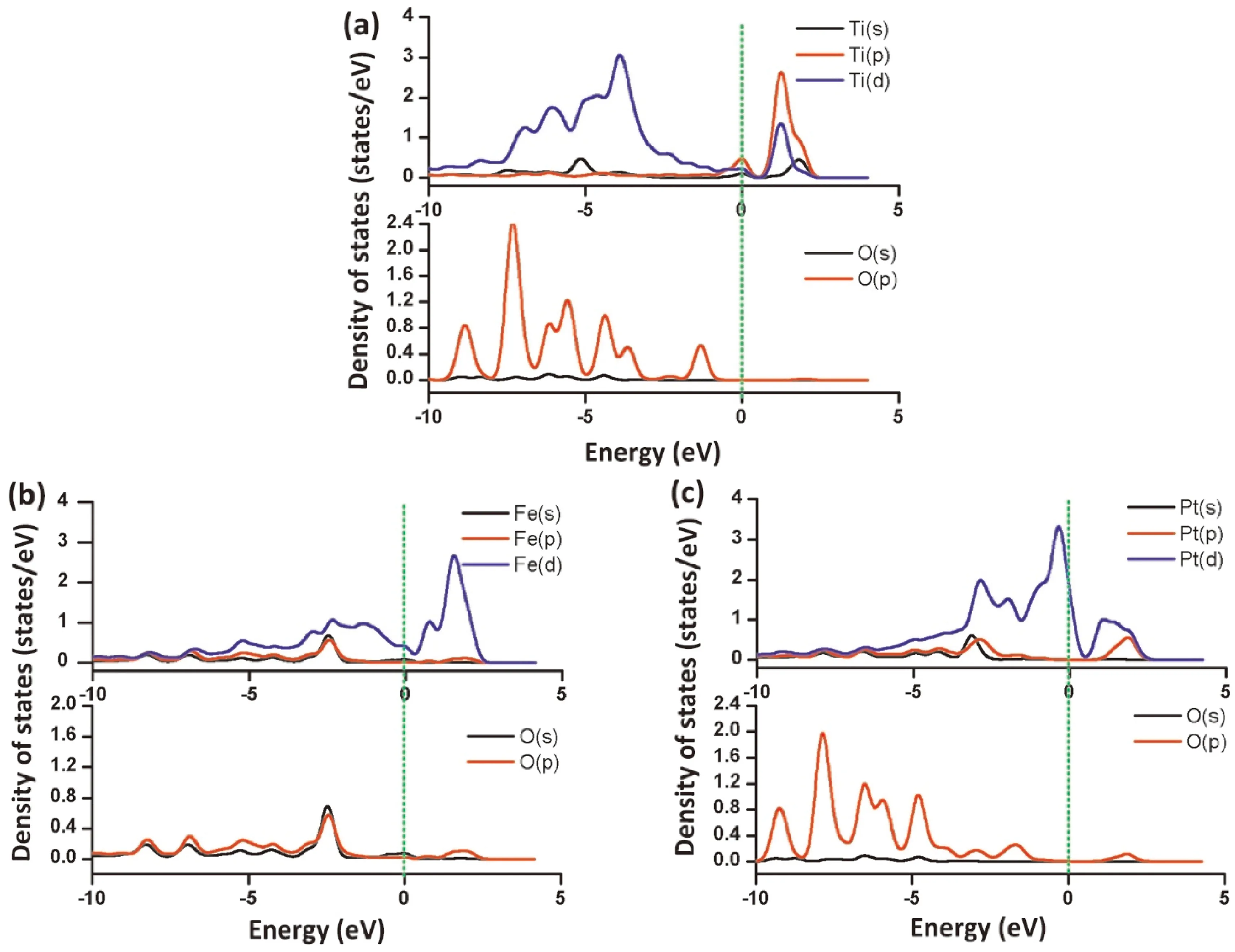
图 5 分态密度图. (a) 在TCDF-Ti-G模型中Ti和O原子; (b) 在TCDF-Fe-G模型中Fe和O原子; (c) 在TCDF-Pt-G模型中Pt和O原子.Fig. 5 Partial densities of states (PDOS) for (a) the Ti and O atoms in the TCDF-Ti-G model; (b) the Fe and O atoms in the TCDF-Fe-G model; (c) the Pt and O atoms in the TCDF-Pt-G model.
3.3 差分电荷密度分析
电荷密度的结果进一步展示了TCDF和石墨烯之间的相互作用, 图6表明了TCDF与石墨烯表面差分电荷密度的结果. 差分电荷密度表明了电荷密度在吸附过程中的变化, 是由整个吸附体系的总电荷密度(ρTCDF+surface)减掉TCDF分子(ρTCDF)和石墨烯表面(ρsurface)的电荷密度之和, 如公式(2)所示:
ρads=ρTCDF+surface-(ρTCDF+ρsurface)
(2)
红色区域代表电荷聚集的区域, 蓝色区域代表电荷减少的区域[16]. TCDF吸附于本征及掺杂石墨烯表面的差分电荷密度如图6所示. 图6a表明TCDF与本征石墨烯作用时, 电荷减少主要发生在与TCDF中Cl原子相互作用的石墨烯表面, 说明石墨烯表面电子转移到了TCDF中Cl原子中; 电荷增加主要在TCDF中-CH2周围的石墨烯表面, 说明TCDF中-CH2电子转移到了石墨烯表面, 因此TCDF和本征石墨烯表面应该发生了电荷转移, 它们之间形成了C-Cl…π和C-H…π相互作用. Kumar[29]研究了X-H…π相互作用, 结果表明电荷从-CH流向苯环, 使苯环电子密度增加, 从而证明了C-H…π非共价相互作用的存在. Sangeetha等人[30]表明2,4-二胺-6-苯-1,3,5-三嗪环/4-氯苯甲酸能够稳定存在, 是因为它们之间存在着弱的C-Cl…π非共价相互作用.

图 6 等值面为0.20 e Å-3 的电子差分密度的三维图. TCDF吸附在(a) 本征石墨烯; (b) Ti掺杂石墨烯; (c) Fe掺杂石墨烯; (d) Pt掺杂石墨烯表面.Fig. 6 Three-dimensional plots of the electron density difference with an isovalue of 0.20 e Å-3: TCDF adsorptions on the (a) intrinsic graphene; (b) Ti-doped G; (c) Fe-doped G; (d) Pt-doped G surfaces.
Ti和Fe掺杂的石墨烯表面(图6b和图6c), 金属原子Ti和Fe周围是蓝色, 表明它们失去了电子, 而TCDF中O原子周围为红色, 表明O原子得到了电子, 因此, 它们之间形成了Ti-O和Fe-O共价相互作用. 在Pt掺杂石墨烯表面, 它们之间没有明显的颜色变化, 因此电荷转移量也较少(图6d), 相互作用也较弱.
4 结 论
了解剧毒物质二噁英TCDF在材料表面的吸附机理对于开发各种环境中有毒物质去除和检测的新方法起着非常重要的作用. 本研究利用密度泛函理论方法探索了用本征石墨烯, Ti, Fe和Pt掺杂石墨烯作为二噁英污染物TCDF分子的潜在高效传感器的可能性. 结果表明, 四种石墨烯均能有效去除TCDF分子, 其中, Ti和Fe掺杂原子的存在进一步增加了石墨烯基底对TCDF分子的敏感性. 此研究结果有望为检测和去除剧毒物质二噁英污染物提供理论指导和帮助.
