铂催化3-炔-1-醇分子内加氢烷氧基化反应的理论研究
2021-09-16张兴辉
张兴辉
(兰州文理学院 化工学院, 兰州 730010 )
1 引 言
烯醇醚由于π键的富电子特性是非常有用的官能团,可以在一系列转化反应中实现独特的反应性[1]. 烯醇醚的合成方法有多种,包括金属催化的方法,例如醇/烯基卤化物的交叉偶联,烯丙基醚异构化和炔烃加氢烷氧基化反应等,尤其后一种反应方式更加直接、方便,在该反应中主要考虑添加的区域选择性[2-5]. 鉴于天然产物和生物活性分子中可能普遍存在烯醇醚,这些分子主要来源于选择性的内环或外分子内环化反应,因此选择性地实现这些过程的方法具有很好的潜在研究价值[6]. 已有研究报道显示铂催化炔丙基醚能够生成α,β-不饱和卡宾中间体,表明这些卡宾可以进行环加成,氢迁移和乙烯基亲核加成,而且 6-内-环化可能对于5-外-环化形成竞争机制[7-9]. 金属催化的分子内炔烃加氢烷氧基化反应中的区域选择性已经被实验研究,但决定因素相对复杂. 合成机理的差异可以得到不同的产物,在π活化过程中,末端炔烃通常会附着在金属催化剂上,驻留在乙烯基金属中间体的未取代碳上[10]. 随后,Liu和De Brabander等人[11]研究表明,可以通过特定催化剂中间体的路易斯侧基来调节烷基体系中的区域选择性. 诸如催化剂选择和空间环境等其他方面也是主要的影响因素. 研究显示其他非金属和金属(金和钯)催化的加氢烷氧基化反应中也体现着一定的选择性,表明其存在着电子偏压[12]. 最近,Costello等人[13]报道了在甲苯溶液中,在没有其他配体和添加剂的条件下,[(C2H4)PtCl2]2催化苯基3-炔基-1-醇的环加成反应,在此扫描中观察到明显的环化方向性分离(图1). 为了进一步研究该反应的具体反应机理和区域选择性,本文以苯基3-炔-1-醇为反应物,以PtCl2为催化剂对铂催化的分子内加氢烷氧基化反应过程进行密度泛函计算研究,研究得到的反应机理和区域选择性计算数据有助于更好地理解铂催化的此类反应的微观状态,并对此类有机反应的合成具有很好的指导和借鉴意义.
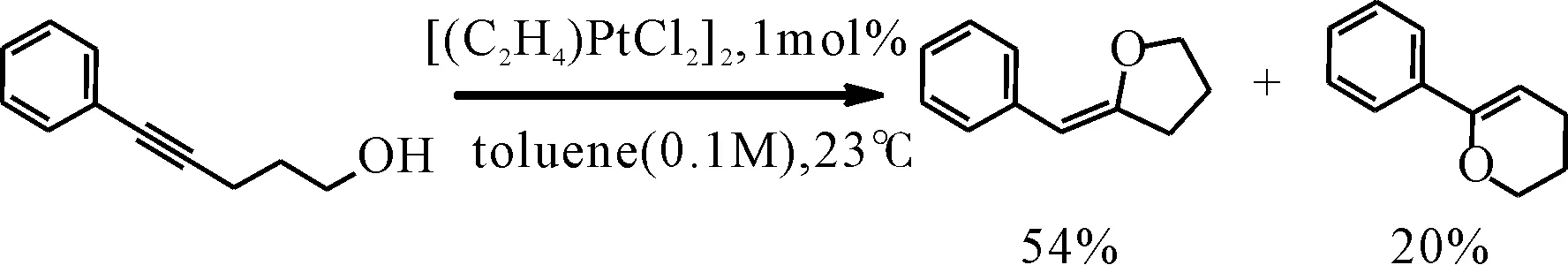
图1 铂催化的分子内加氢烷氧基化反应的选择性Fig. 1 Regioselectivity in Pt-catalyzed intramolecular hydroalkoxylation
2 计算方法
通过使用M06-2X方法对反应体系进行了DFT计算,在气相中完全优化得到所有物种的几何构型和能量,这种计算方法可以提供准确的能量并广泛地应用于金属催化反应的许多理论研究[14-16]. 在计算中,C,H,O和Cl采用6-311G(d,p)基组,Pt原子选择SDD雁势基组进行构型优化. 所有驻点结构都在相同理论水平上计算了频率分析来进行表征,中间体没有虚频率而过渡态有唯一虚频,并通过零点能(ZPE)对相对能量进行了校正. 各过渡态结构通过内禀反应坐标IRC计算验证TSs结构正确连接两个对应的能量最小值中间体结构. 基于实验条件,以甲苯作为溶剂,使用SMD溶剂化模型对气相优化的结构在M06-2X / 6-311++G (d, p) / SDD 理论水平上进行了单点能计算. 本文讨论所用的能量均为考虑了溶剂化的自由能,所有几何构型的优化以及能量计算均使用高斯09软件包来完成.3D分子结构是借助CYL view软件直接从计算输出构型生成的.
3 结果与讨论
为了详细研究铂催化的炔醇化合物分子内加氢烷氧基化反应的机理和区域选择性,根据实验研究报道,使用铂金属络合物RC(图1中炔醇反应底物骨架与催化剂PtCl2的铂原子相互作用被活化)作为催化反应的前驱配合物. 图2给出了优化的铂金属配合物结构RC的HOMO和LUMO前线分子轨道图和静电势图. 静电势图显示出分子体系中存在较强的电荷分离现象,分子结构中的氧和氯原子有较强电负性,其他结构基团均存在电正性,尤其是羟基的H原子最强. 结构RC的HOMO显示出Pt原子与炔醇中炔烃π键之间的相互作用,Pt与炔基形成的三角形骨架上的电子云分布较大,而LUMO显然能够看到炔基的π轨道和Pt的d轨道,羟基上的电子分布极其少,则炔基就是分子RC中的最大活性位点. RC分子中的HOMO和LUMO轨道的能量差相对较小,为0.1820 eV. 亲核基团羟基中的氧原子由于具有较强的电负性(O: -0.564 e),更容易进攻CR分子中的炔基π键进行环化. 图3给出了从反应配合物RC开始铂催化的分子内加氢烷氧基化反应的势能面图,图4提供了势能面上各驻点的几何构型及关键几何参数. 在RC中,与Pt原子形成等腰三角形三元环骨架结构,其中Pt-C1和Pt-C2键长分别为0.2092和0.2094 nm, C1-C2也被拉长为0.1259 nm,碳碳三键已经被初步活化.
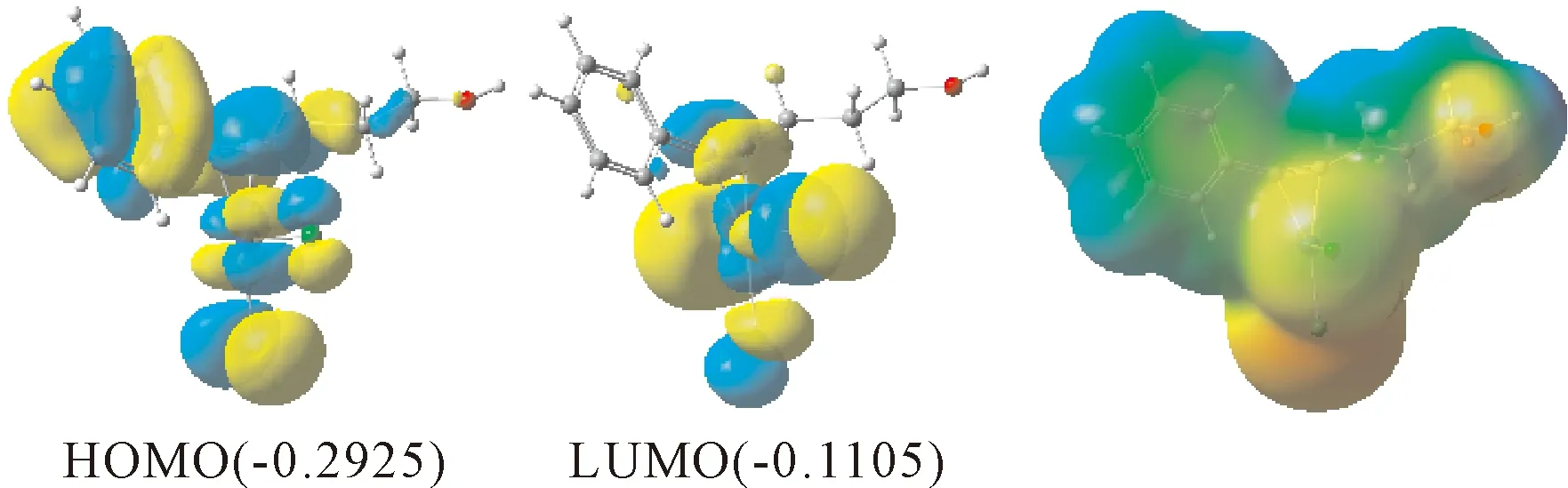
图2 RC初始反应物分子的前线分子轨道和静电势图Fig. 2 Frontal molecular orbitals and electrostatic potential diagram of the molecular structure of RC reactant
在图3中,沿着反应物RC,羟基氧原子作为亲核试剂分别进攻炔基中的两个不同碳原子进行分子内五元或者六元环化过程,形成了两个不同的竞争性反应通道. 首先,通过过渡态TS1羟基氧原子进攻C2得到五元环中间体IM1,TS1拥有唯一一个虚频率为414.3 i cm-1,虚频对应的振动模式主要体现在O-C2上,Pt-C2键也趋于断裂向C1转移,IRC计算也证实过渡态TS1连接着初始反应配合物和对应中间体. 由图3能够看出,这一步反应的活化自由能相对于RC为30.8 kJ / mol,中间体IM1相对于反应物的能量为-5.2 kJ / mol. 在中间体IM1中,新O-C2键已完全形成,其键长为0.1504 nm,PtCl2也完成了向C1的转移,Pt-C1键比反应物缩短了0.0178 nm. C1和C2原子的杂化方式也由sp转化为sp2. 随后,IM1通过一个氢转移过渡态TS2(虚频为1126.5 i cm-1)完成了向C1原子的氢迁移. 在TS2中,O-H和C1-H键长分别为0.1098和0.1750 nm,Pt原子又开始向C2原子迁移. 从IM1到TS2也是该反应通道的决速步骤,其活化自由能为139.0 kJ / mol,中间体IM2相对于IM1放热量为113.1kJ / mol,表明中间体IM2稳定性比较高. 在IM2中,五元环产物骨架已初步形成,催化剂与C1和C2原子再次形成同时相互作用,Pt-C1和Pt-C2键长分别为0.2063和0.2201 nm. 最后,中间体吸收121.3 kJ/mol得到实验合成的五元环产物并释放出PtCl2催化剂. 相对于反应物而言,整个反应路径几乎无热效应,吸热量仅为3.0 kJ / mol.
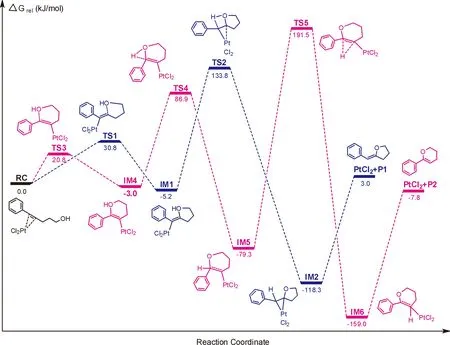
图 3 铂催化的分子内加氢烷氧基化的势能剖面图Fig.3 The potential energy surface profile of Pt-catalyzed intramolecular hydroalkoxylation

图 4 铂催化的加氢烷氧基化反应势能面上各驻点几何构型及参数(键长/ nm)Fig. 4 Geometries and parameters of the potential energy surface of Pt-catalyzed hydroalkoxylation(bond length / nm)
然而,若羟基氧原子选择性进攻另一个炔基C原子形成另外一个竞争反应通道,即通过过渡态TS3的6-内切环化就可以得到六元环中间体IM4. 第一步对应于五元环化过程(TS1)其活化自由能降低了10.0 kJ / mol,反应更加容易. 在TS3中,O-C1键长为0.2056 nm,Pt-C1键完全断裂并与C2原子的作用力进一步增强,Pt-C2键比反应物缩短了0.0145 nm. 随后,沿着IM4通过过渡态TS4完成了羟基上氢原子向C1原子的第一次迁移,这一步的活化自由能相对于中间体IM4是89.9 kJ / mol. TS4的唯一虚频为1607.5 i cm-1,O-H和C1-H键长分别为0.1243和0.1305 nm. 反应接着进行二次氢迁移,由过渡态TS5使得H原子从C1转移到C2原子上,这一步活化能相对于中间体IM5为270.8 kJ / mol,该步骤也是第二个通道的决速步骤,具有比较高的活化能. 中间体IM6最终吸收151.2 kJ / mol就可以得到六元环选择性产物P2,并释放出催化剂.
对比两个反应通道,相对于反应物RC而言,生成五元环P1的反应路径具有相对较低的活化能,其值为133.8 kJ / mol,生成六元环产物P2的活化自由能比P1高了57.7kJ / mol,表明合成产物以P1为主,但两个通道相对较小的能量差,说明后者通道会对五元环路径构成竞争反应,也就是会有部分六元环产物生成. Costello等人的实验研究显示,在相同的实验条件下,铂催化3-炔-1-醇分子内加氢烷氧基化反应得到两种产物的产率分别为54%和20%. 其中五元环为主产物,这与本文的计算研究结果完全一致,计算结果也对该反应的实验结果给予了很好理论解释.
4 结 论
通过M06-2X计算方法详细研究了铂催化3-炔-1-醇分子内加氢烷氧基化反应的反应机理和区域选择性. 铂催化的整个反应过程有两个关键步骤,即羟基氧原子进攻炔烃碳原子的环化过程和氢迁移机理得到目标合成产物并释放催化剂PtCl2. 计算研究表明,对于选择性进攻炔基不同的碳原子形成的竞争机制,氢迁移步骤具有相对较高的活化能,从反应动力学上更倾向于形成五元环产物的反应通道,其活化自由能为133.8 kJ / mol. 两个反应路径的能量差为57.7 kJ / mol,存在一定的选择性但也构成了一定的竞争性,即以五元环产物为主,也伴随着生成了部分的六元环产物. 计算研究结果与实验合成完全一致,并给予了详细的机理分析和选择性解释,为进一步研究金属催化的加氢烷氧基化合成反应提供了有力支撑.
