单原子Bi吸附对BiOI光催化性能的影响
2021-09-16朱超涌
李 庚, 朱超涌
(四川大学 原子与分子物理研究所, 成都610065)
1 引 言
随着人类社会发展对能源日益增长的需求,及日趋严重的环境污染问题,利用清洁能源成为人类主要的研究方向之一. 半导体光催解剂因其太阳能转换效率高、环境污染低而备受关注[1-2]. 近年来,研究发现了许多具有很高光催化效率的半导体材料,如ZnO[3]和TiO2[4-5],其中,TiO2因其环保、成矿完整、成本低、无毒等优点,成为研究最广泛的光催化剂[6-7]. 然而,TiO2的带隙较宽(3.2 eV),导致TiO2对太阳能的吸收率较低(<4%),此外,较高的光生电子和空穴复合率,也极大地限制了TiO2的实际应用[8]. 学者还研究了其它由可见光驱动的高效光催化剂,如CdS[9]、BiOBr[10-11]、BiOI等,其中BIOI因其无毒、环保等优势也备受关注[13],BiOI被认为是最有潜力的光催化剂之一[12].
BiOI的晶体结构表明 [Bi2O2]2+层镶嵌在两层 I2-中间,其结构使其具有狭窄的波段间隙,通常用作可见光催化剂,可以在可见光照射下激活[14-15]. Dai等人[16]详细研究了 BiOI 晶体的电子结构和光学特性,Huang等人[17]使用 DFT 方法计算了 BIOX(X = F、Cl、Br、I) 的电子结构和光学特性,Long等人[13]合成多孔分层 BiOI 纳米结构,罗丹明B(RHB)的去除率达到 98.7%,表明其形态和结构对BiOI的光催化性能有重要影响. 前期研究表明光催化发生在BiOI表面,裸露的晶体表面性质在光催化效率方面起着重要作用. Ye等人[14]合成了具有(001)和(100)暴露面的BiOI,实验表明BIOI(001)对CO和CH4的产量分别为5.18 μmol h-1g-1和1.78 μmol h-1g-1,而BIOI(100)对CO和CH4的产量分别为1.52 μmol h-1g-1和1.50 μmol h-1g-1. Shan等人[15]通过简单的化学转换技术成功制备了BiOI,实验表明,具有(001)暴露面的BiOI有利于增强可见光催化,此外,Zhang等人[18]使用 DFT 计算来研究 BiOX (X = F、Cl、Br、I)(001)、(110)和(010)面的光催化特性,发现表面为X原子的 (001) 面具有极高的热力学稳定性,Kong等人[19]发现,表面为I原子的BIOI(001)(1I-BiOI)的表面能最低.
研究表明虽然 1I-BiOI(001)的光催化性能高于BiOI块体,但仍达不到应用的要求,对此有研究人员提出一些方法来解决光催化性能差的问题,如掺杂、引入缺陷以及吸附. 金属/半导体复合材料可以显著提高半导体材料的光催化效率[20-23],主要原因是金属和半导体材料在接触面形成肖特基势垒,阻碍电子空穴复合;其次是金属和半导体之间的内建电场,有利于电子-空穴分离;同时,在紫外光和可见光条件下,半金属产生等离子体共振(SPR)效应将热载流子从金属转移到BIOI[24-25],从而增强半导体复合材料载流子的分离效率[26-28]. 由成本低廉、高效、容易合成的Bi和半导体组成的金属/半导体材料被广泛使用,如Bi/BiOBr[29], Bi/BiOCl[30], Bi/Bi2O2CO3[31], Bi/BiOI,. 相比于普通半导体,加载Bi后形成的金属/半导体材料能够吸收更多的可见光,且带隙更小. Chang等人[32]合成出一种三维Bi/BIOI复合材料,并将其应用于 BPA 的降解,结果表明制备的 Bi/BiOI 的光催化降解效率高于纯 BIOI;Li等人[33]合成了含有O空位的 Bi/BiOI用来去除NOx,与纯 BiOI 相比,获得的 Bi/BiOI 显示更好的NOX(NO和NO2)去除效率;Liu等人[34]通过电化学沉积和氢还原作用合成Bi/BIOI异构薄膜,光电流密度比BiOI 增加6.3 倍,扩大了可见光吸收范围,提高光电化学活性;此外,Bi/BiOI/X三层异质结催化剂也有研究[35],而双金属和石墨烯的同步耦合同样使BiOI的光催化效率增强[37]. 为了从原子排布和电子结构层面系统研究Bi在BiOI表面负载的微观机理,本文构建了单原子Bi吸附在1I-BiOI (001) 表面的模型,计算研究了不同体系的电子结构和光学性质,探究Bi原子吸附对提高BiOI催化性能的机理及过程.
2 计算方法和模型
基于密度泛函理论(DFT)、采用第一性原理计算程序Vienna ab initio simulation package (VASP)完成所有的计算研究[38-40]. 使用Perdew-Burke-Ernzerhof ( PBE ) 的广义梯度近似(GGA)描述交换势和关联势,采用经验修正方法 (DFT-D2) 来修正范德瓦尔斯相互作用[41, 42]. 使用的平面波截断能量为490 eV,能量收敛标准为5.0×10-7eV,3 × 3 × 1 K点采样布里渊区.
BiOI属于P4/nmm空间群,单胞包含四个原子,其晶格参数为a=b= 3.984 Å,α=β=γ= 90°[37],如图1(a)所示. 使用 PBE计算优化后的结构为a=b= 3.999 Å,扩展得到2×2×2 超胞,由此构建表面为I原子的(001)面结构模型,如图1(b)所示. 在1I-BiOI表面添加一个Bi原子得到吸附Bi的1I-BiOI体系模型,其中三个不同的吸附位点是Z、B和H,如图1(c)所示. 单原子 Bi 在 Z、B 和 H 位点被吸附,表示为Bi-Z, Bi-B和Bi-H吸附体系. 在 Z 方向添加了 15 Å 的真空层,如图1(d-e)所示.
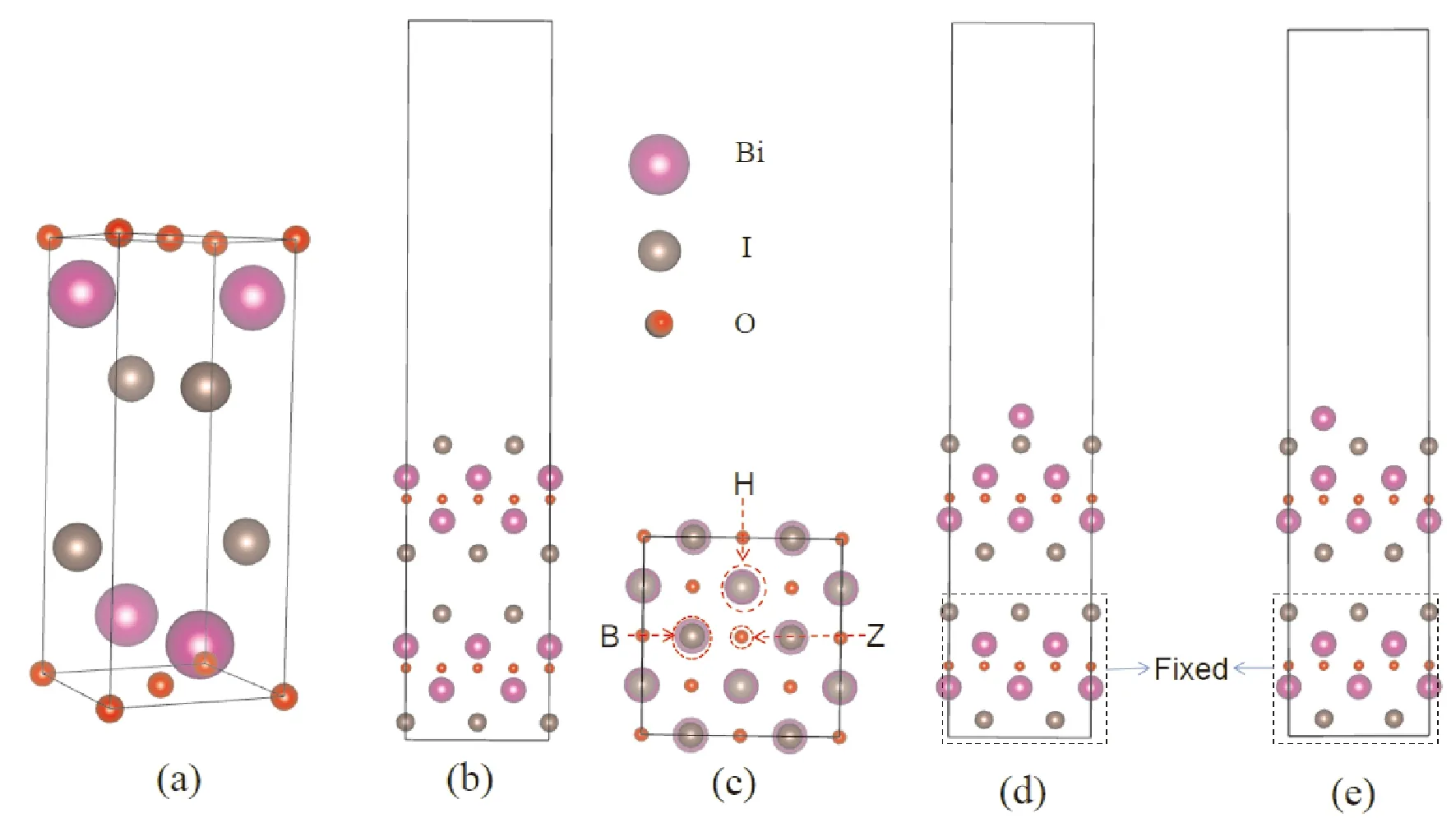
图1 (a) BiOI体晶体结构, (b) 1I-BiOI表面的晶体结构,(c)1I-BiOI 的俯视图,B、Z 和 H表示三个不同的吸附位点. 单原子Bi吸附在BiOI (001)上,(d) Z、H和 (e) B位点的侧视图Fig. 1 Crystal structures of (a) BiOI bulk and (b) 1I-BiOI surface. (c) Schematic representations of top view of 1I-BiOI, B, Z and H represent three different adsorbed sites. Schematic representations of side view of monatomic Bi adsorbed at the (d) Z, H and (e) B sites on the BiOI (001) surface and fixed the underlying atoms.
3 结果和讨论
3.1结构优化和吸附能
优化后的Bi-X (X = Z, B, H) 的晶体结构如图2所示. 计算结果表明优化前后吸附原子Bi与周围原子的位置发生了变化,优化前吸附原子Bi与1I-BiOI表面的距离为1.614 Å. 优化后,Bi-B体系吸附原子Bi与1I-BiOI表面的距离约为1.922 Å,而Bi-Z和Bi-H体系吸附原子Bi与1I-BiOI表面的距离分别为1.978 Å和2.879 Å, 如表1.

图2 优化后的 (a) Bi-Z,(b) Bi-B和 (c) Bi-H的晶体结构Fig. 2 Optimized crystal structures of (a) Bi-Z, (b) Bi-B and (c) Bi-H
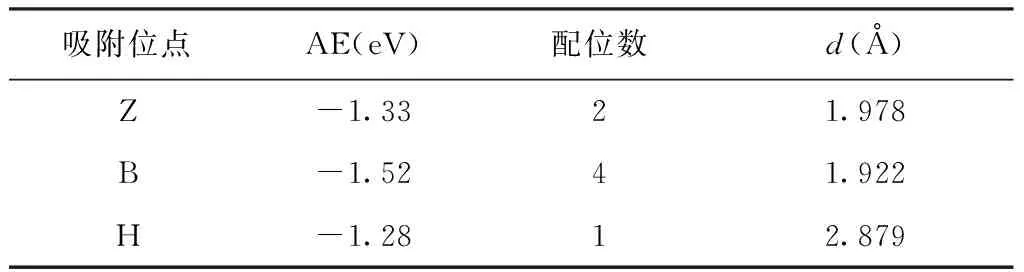
表1 1I-BiOI表面Z、B和H位点上的吸附能(AE)、配位数和吸附原子到表面的距离d.
为了研究吸附结构的稳定性,进一步计算吸附体系的吸附能,吸附能(AE)计算公式可表示如下:
AE=ETot-Esub-nEi
(1)
其中,ETot是原子吸附后体系的总能量,Esub是原子吸附前系统的总能量,Ei是吸附原子的能量,n是吸附原子个数. 在此研究系统中,Ei指单原子Bi的能量,n值为1. 1I-BiOI表面Z、B和H位点的单原子Bi吸附能如表1. 所得结果说明吸附能为负值,即单原子 Bi 很容易在 1I-BiOI表面吸附. 通过比较不同吸附位点的吸附能,发现Bi-B系统的吸附能低于Bi-Z系统,表明Bi原子更倾向于B点位吸附. 计算研究了三个不同吸附位点吸附原子的配位数(配位数是指晶体中原子周围最近的原子个数),分别为2(Z位)、4(B位)和1(H位). 结果表明:吸附能和吸附几何构型均与Bi的配位数有关,Bi与1I-BiOI表面的距离随着Bi配位数的增加而减小;同时,配位数越大,吸附能越低.
3.2 电子结构
计算得到了1I-BiOI体系的能带结构,如图3(a)所示. 计算得到的带隙为1.54 eV,与实验中测得的BiOI薄膜禁带宽度1.6 ± 0.1 eV、1.77 eV[5]基本吻合. 进一步计算研究了Bi-Z、Bi-B和Bi-H不同吸附位点体系的带隙,计算结果分别为0.81 eV、1.36 eV和1.11 eV,其VB和CB向低能量方向移动,使吸附体系成为氧化性能增强的n型半导体,如图3(b-d)所示. 对于Bi-Z和Bi-H吸收体系,其带隙减小,有利于低能段可见光的吸收. Bi-B吸附体系不仅使带隙减小,而且在导带和价带之间产生了明显的杂质能级,使价带电子也可以利用能量较低段的光辐射吸收,先跃迁到杂质级,再跃迁到导带,从而明显提高光能利用效率. 同时,杂质能级可以作为光诱导载流子的捕获中心,有利于提高光诱导载流子的分离效率[5]. 在实验中,Bi金属加载到半导体上后,带隙减小,这与我们计算的每个位点上吸附Bi原子后带隙减小的结果一致[29-31, 36].
此外,计算了1I-BiOI、Bi-Z、Bi-B和Bi-H体系的投影态密度(PDOS)和总态密度(TDOS) (图4). 从图4 (a-d)可以看出,1I-BiOI和吸附体系的VBM主要由I 5p态贡献,1I-BiOI体系的CBM是由Bi 6p态贡献的. 对于Bi-Z、Bi-B和Bi-H吸附体系的CBM,Bi 6p态贡献较大,I 5p和O 2p态贡献较小. 如图4 (c)所示,Bi 6p、I 5p和O 2p态都对杂质能级有贡献. 在B位点的吸附体系中,吸附原子Bi与表面I、O原子发生杂化,形成杂质能级,杂质能级作为光电子的捕获中心,可以抑制光诱导的电子空穴复合,提高材料的光催化效率.
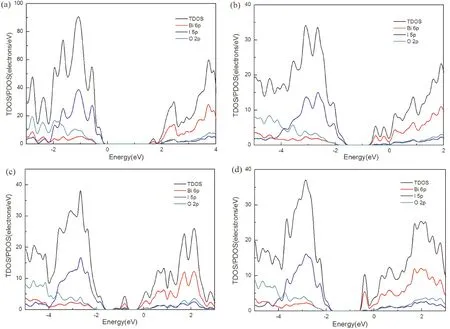
图4 (a) 1I-BiOI (b) Bi-Z (c) Bi-B和 (d) Bi-H体系的计算投影态密度(PDOS)和总态密度(TDOS). Fig. 4 Calculated projected densities of states (PDOSs) and total densities of states (TDOSs) of (a) clean 1I-BiOI (b) Bi-Z (c) Bi-B and (d) Bi-H systems.
3.3 电荷转移和功函数
为了探究Bi-X (X = Z, B, H)体系中电荷转移和分离的性质,计算了体系的平面平均电荷密度的差值,采用平面平均电荷差密度分析界面电荷分布,定义为:
Δρ=ρBi-X-ρBi-ρBiOI
(2)
ρBi-X为Bi-X系统的平面平均电荷密度,ρBi和ρBiOI分别为单原子Bi和1I-BiOI体系的平面平均电荷密度. Bi-X吸收系统Z方向的平面平均电荷密度,图5给出了不同吸附位点体系的电荷分布,正数表示电荷积累,负数表示电荷耗尽,图5(a)所示的Bi-Z吸附体系中,吸附原子Bi的有效电子耗散量约为0.004, 1I-BiOI最上层I原子以上的有效电子积累量约为0.0045,图5 (b-c)所示Bi-B和Bi-H吸附体系也有类似的结果. 表明电子从Bi吸附原子转移到1I-BiOI表面的I原子,这与Bi原子加载到半导体上后,电子从Bi向半导体表面转移的实验结果一致.

图5 (a) Bi-Z (b) Bi-B (c) Bi-H系统的平面平均微分电荷密度 Δρ (z).Fig.5 Planar-averaged differential charge densities Δρ(z) of (a) Bi-Z (b) Bi-B (c) Bi-H systems.
功函数是描述电荷转移的重要参考参数. 功函数(φ)的定义如下:
φ=Evac-Ef
(3)
其中Evac表示真空中靠近表面的静止电子的能量,Ef为材料的费米能级. 计算得到的1I-BiOI和不同吸附体系的功函数如图6所示,1I-BiOI系统的功函数为6.07eV,单原子Bi原子被吸附后,Bi-Z、Bi-B和Bi-H系统的功函数分别为4.58、4.61和4.67eV. 相比较,Bi原子吸附后体系的功函数均减小. 其可能的原因是: 1I-BiOI表面最上层的I原子比吸附原子Bi具有更强的电负性,吸附原子Bi将电子从吸附原子Bi转移到1I-BiOI表面最上层的I原子,从而在带正电的Bi原子和带负电的I原子之间产生内建电场. 另外,内建电场会抵消正极[Bi2O2]2+与负极I-之间的部分内建电场,导致功函数减小. 吸附体系功函数的减小,表明单原子Bi吸附在1I-BiOI表面更利于转移电子. 同时,由如图6 (b-c)可知Bi-Z、Bi-B和Bi-H体系均有较小的电势降,说明电子从吸附原子Bi表面转移到1I-BiOI表面,与前面平面平均电荷密度分析的结果一致.
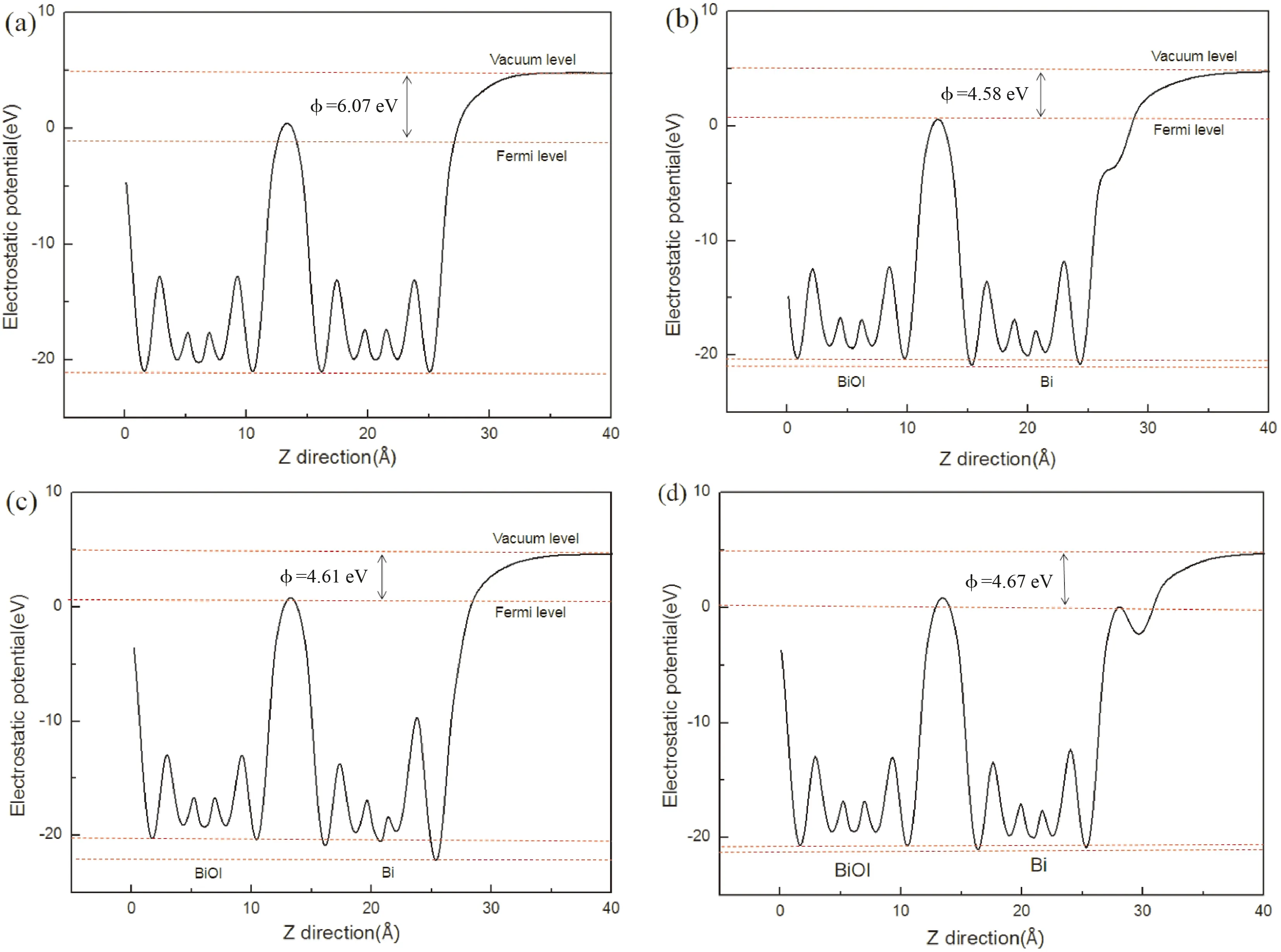
图6 (a) 1I-BiOI (b) Bi-Z (c) Bi-B和 (d) Bi-H体系沿Z方向的计算静电势. Fig. 6 Calculated electrostatic potentials along Z direction of (a) clean 1I-BiOI (b) Bi-Z (c) Bi-B and (d) Bi-H systems.
3.4 光学特性
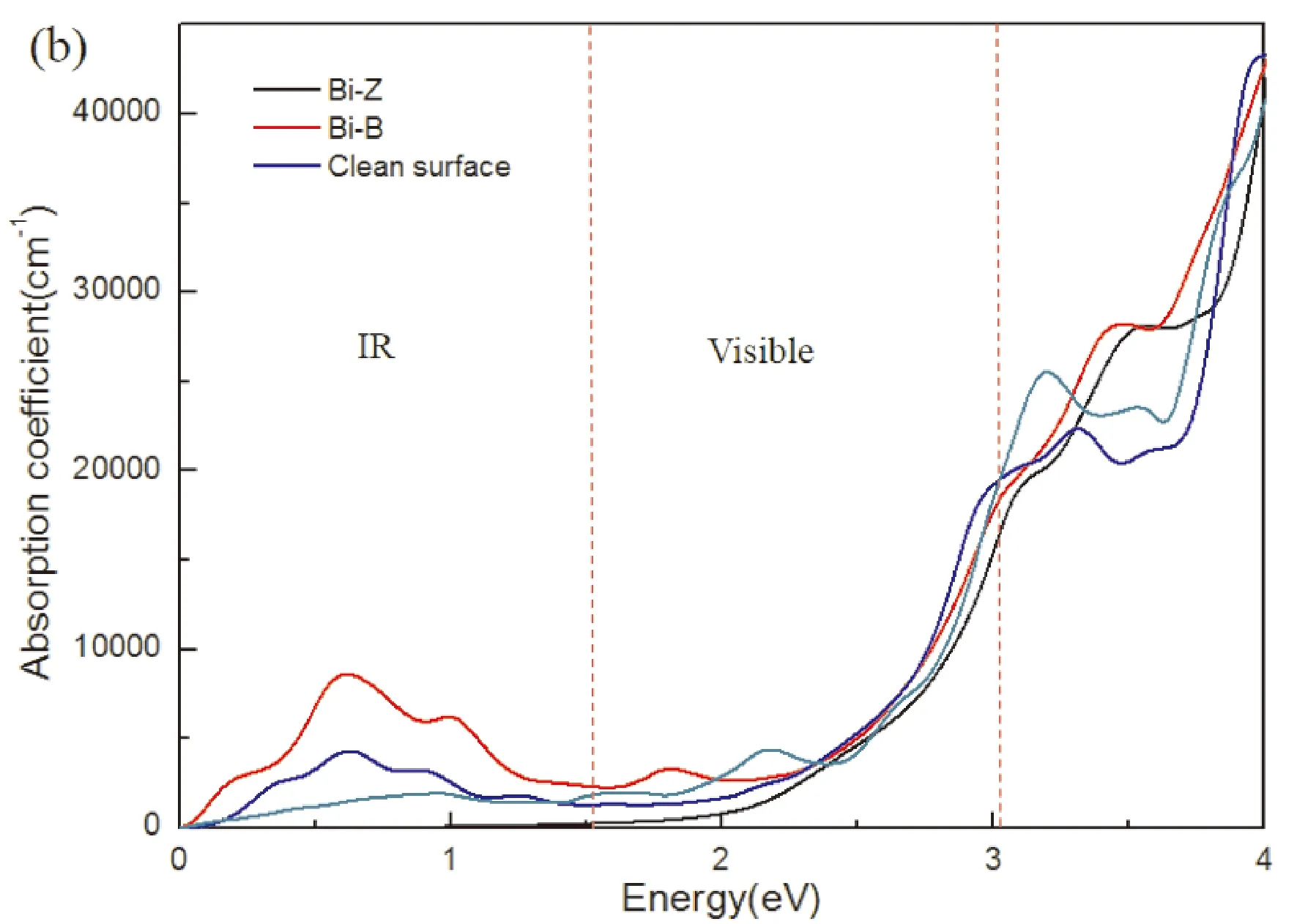
材料的光学性能是光催化剂的一个极其重要的参数. 为了探究单原子Bi吸附在1I-BiOI表面对光学性能的影响,分别计算1I-BiOI、Bi-Z、Bi-B和Bi-H体系的吸收光谱,图7所示. 吸收系数表示为:
(4)
其中ε1(ω)和ε2(ω)分别是介电函数的实部和虚部,它们取决于光频率(ω),虚部ε2(ω),与光催化材料的光吸收密切相关,计算结果如图7 (a)所示. 结果表明,BiOI的光吸收能力较差,对可见光(1.6 eV 图7 (a) 介电函数的虚部ε2(ω),(b) 1I-BiOI,Bi-Z, Bi-B和Bi-H体系的光学吸收光谱. Fig. 7 (a) Imaginary part ε2(ω) of the dielectric functions and (b) the optical absorption spectra of clean 1I-BiOI surface, Bi-Z, Bi-B and Bi-H systems. 本文采用第一性原理计算方法研究了单原子Bi吸附在1I-BiOI表面的结构、电子和光学性质. 研究结果表明,与BiOI相比,Bi/BiOI吸附体系不仅具有更好的结构稳定性,而且具有更好的可见光利用率. 当单原子Bi吸附在1I-BiOI表面时,形成n型半导体,增强了氧化性能. 平面平均差分电荷密度分析表明,电荷从吸附原子Bi转移到1I-BiOI表面. 与1I-BiOI体系相比,Bi-B、Bi-Z和Bi-H体系的功函数降低,说明吸附原子Bi与1I-BiOI之间形成了内建电场,加速了电子转移,抑制了光电子-空穴结合. 吸收光谱分析表明,单原子Bi吸附后,Bi/BiOI吸附体系的光吸收增强. 此外, 尽管Bi-Z系统有更大的光吸收, Bi-B中的杂质能级系统可以作为光生电子的捕获中心,抑制电子空穴复合,并且可见光吸收不比Bi-Z体系差,使得Bi-B系统具有更高的光催化效率,进一步提高光催化性能.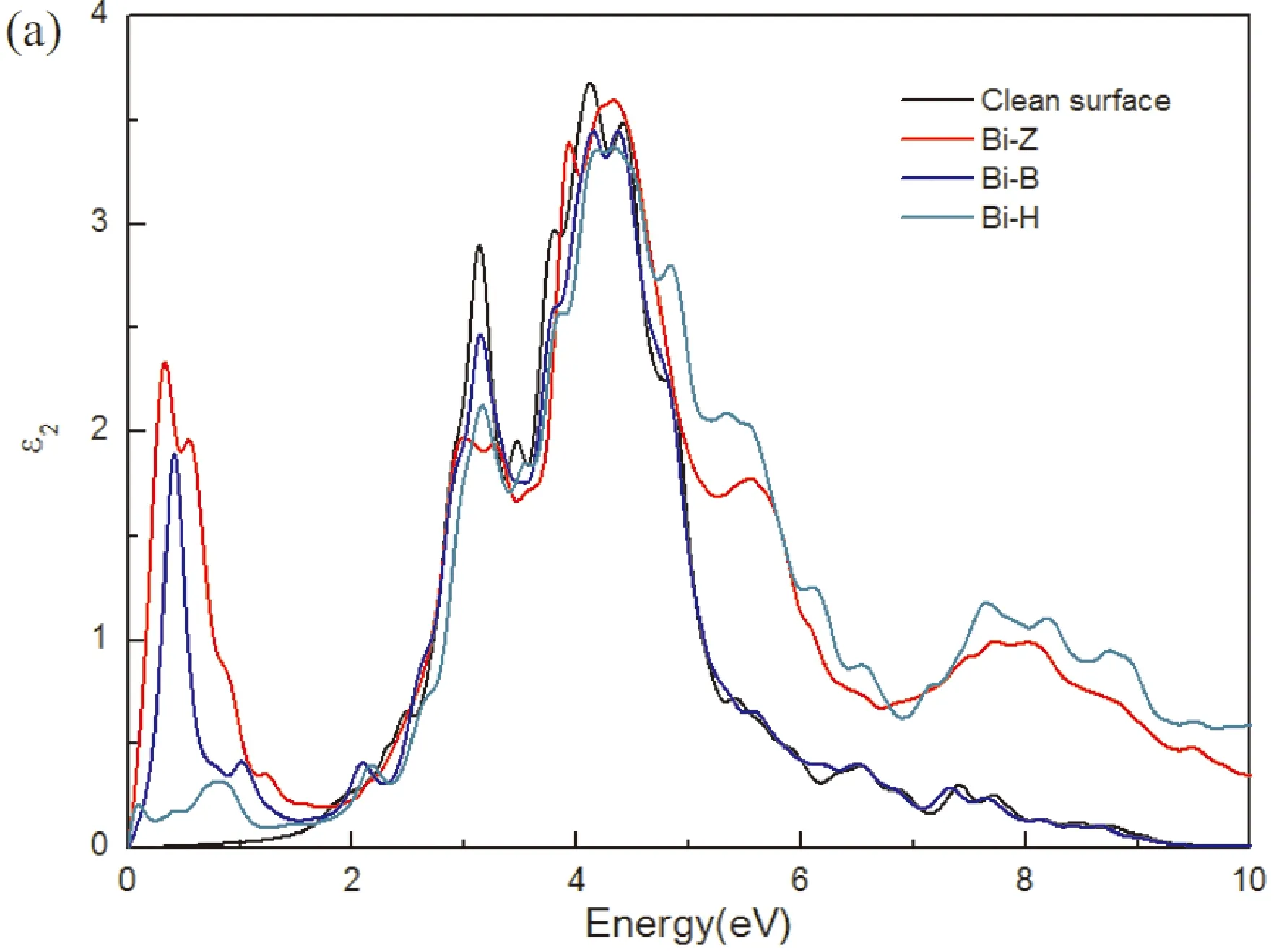
3 结 论
