伴中枢神经系统损害的多发性对称性脂肪瘤病1例报告
2021-09-10刘丽娟李宗树孙宏侠王新丽王明宇
刘丽娟, 李宗树, 孙宏侠, 王新丽, 刘 敏, 王明宇
多发性对称性脂肪瘤病(multiple symmetrical lipomatosis,MSL)是一种原因不明的慢性脂肪异常代谢性疾病,病灶多呈对称性、弥漫性分布于颈背部、胸腹部、四肢近端等部位的皮下、浅筋膜或深筋膜间隙层内。额面部、四肢远端、舌、咽、乳房、阴囊、中枢神经系统及内脏等部位较少累及[1]。本病最早由Brodie BC于1846年首次提出,先后被Madelung OW、Lanois PE和Bensaude R进行归纳和总结,因此也称为马德隆病(Madelung’s disease)或Lanois-Bensaude综合征。MSL在地中海及东欧地区多见,总发病率约为1/40万,30~60 岁中年男性好发,男性发病率最高为女性发病率的30倍[2]。MSL在临床上并不多见,累及中枢神经系统的病例更是少有报导。本篇文章将对我院收治的1例MSL病例进行报导,以增加临床医师对该病的认识。
1 临床资料
患者,男性,41岁。因皮下肿物、尿便障碍33 y、性功能异常20 y、肢体麻木3 y加重9个月,于2020年12月19日收入我院。现病史:患者于33 y前发现双上肢异常肿物,肿物位于双上臂皮下,约黄豆粒大小呈对称性分布,无明显疼痛,当时未在意。近33 y来肿物体积逐渐增大,并累及头颈部、胸背部、骶尾部及双下肢近端。病程中伴有尿便障碍,表现为幼年起常有尿失禁,成年后房事时可有尿液自动排出,8 y前于前卫医院诊断为“神经源性膀胱”行“膀胱造瘘术”,现长期留置导尿管。排便障碍表现为腹泻及房事时便失禁。性功能异常20 y,表现为勃起无力,阳痿。3 y前患者自觉右侧下肢麻木,无头晕、耳鸣,无饮水呛咳、吞咽困难,无肢体活动障碍及言语障碍,无黑蒙及复视,无抽搐及意识障碍。9个月前上述症状加重并出现左侧下肢麻木,表现为行走时明显踩棉花感,站立不稳,向后倾倒。曾行脊椎MRI示“椎管内脂肪瘤”。为求诊治,就诊于我科。既往“先天性骶椎裂、双肾积水”20 y;“右侧面神经麻痹”病史11 y,现遗留右侧面肌痉挛,右侧表情肌活动欠灵活。否认家族遗传性疾病病史。机会性饮酒史20 y,量不多,否认吸烟史。入院查体:头部皮下、颈部、双上肢、胸背部、骶尾部、双下肢近端多处结节样肿块,无压痛、红肿,直径最大者约3×3 cm。神清语明,右侧额纹较左侧稍浅,右侧鼻唇沟浅,示齿口角向左偏斜,余颅神经查体正常。四肢肌力、肌张力正常。双侧共济运动正常。右侧肢体痛觉减退,左侧肢体浅感觉正常。提睾反射未引出,右侧腹壁反射消失。双下肢关节位置觉、音叉振动觉减退,以右侧下肢为著。双侧肱二、三头肌腱反射及膝腱反射对称存在,跟腱反射未引出。病理反射阴性。无项强,克氏征阴性。Romberg征阳性。脊柱无侧弯,活动度正常,无压痛及叩击痛,无隆起及畸形。辅助检查:影像学检查:头部MRI(见图1):双侧外侧裂、颞叶脑沟、小脑沟、四脑室及大脑大静脉池内可见点状T1WI高信号,T2WI高信号,T2-Flair呈高信号,T2-Flair、压脂像低信号;考虑颅内多发脂肪瘤。颈椎+胸椎+腰椎MRI平扫+腰椎增强(见图2):颈椎+胸椎+腰椎脊髓内均可见多发类圆形及条形异常信号,T1WI呈高信号,T2WI呈稍高信号,T2脂肪抑制序列呈低信号;L2-3椎体水平部髓外硬膜下见类圆形异常信号影,较大者大小约36 mm×15 mm,T1WI呈低信号,T2WI呈稍低信号,T2脂肪抑制序列呈高信号,中心部可见结节状低信号,增强扫描呈轻度强化;S3椎体水平部可见类圆形异常信号,大小约18 mm×38 mm,T1WI呈低信号,T2WI呈高信号,T2脂肪抑制序列呈高信号;硬膜囊前缘稍受压,骶椎椎弓见多发不连续,综上考虑颈髓、胸髓、腰髓内多发脂肪瘤,L2-3椎体脊膜瘤;骶管囊肿,隐性骶椎裂。体表肿物及包块彩超(见图3):骶尾部皮下脂肪层可见数个肿物,较大者大小32.8×14.3 mm,考虑脂肪瘤。肌电图:双侧S1水平为主的神经源性损害。实验室检查:全外显子+线粒体基因检测:线粒体基因检测提示存在ND2突变基因。 综合病史、查体及辅助检查结果,临床诊断为多发性对称性脂肪瘤病、骶管囊肿、脊膜瘤、隐性骶椎裂。因经济因素,患者要求在本院保守治疗,经营养神经治疗后,患者双下肢麻木、走路不稳症状减轻。 半年后进行随访,病情暂未进展。
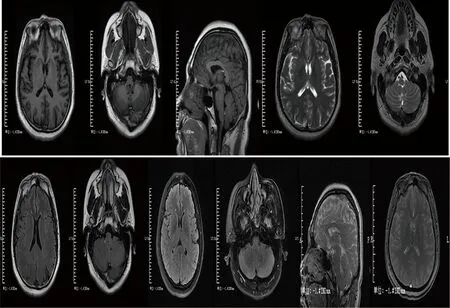
图1 脑沟、脑池及脑室内多发脂肪信号
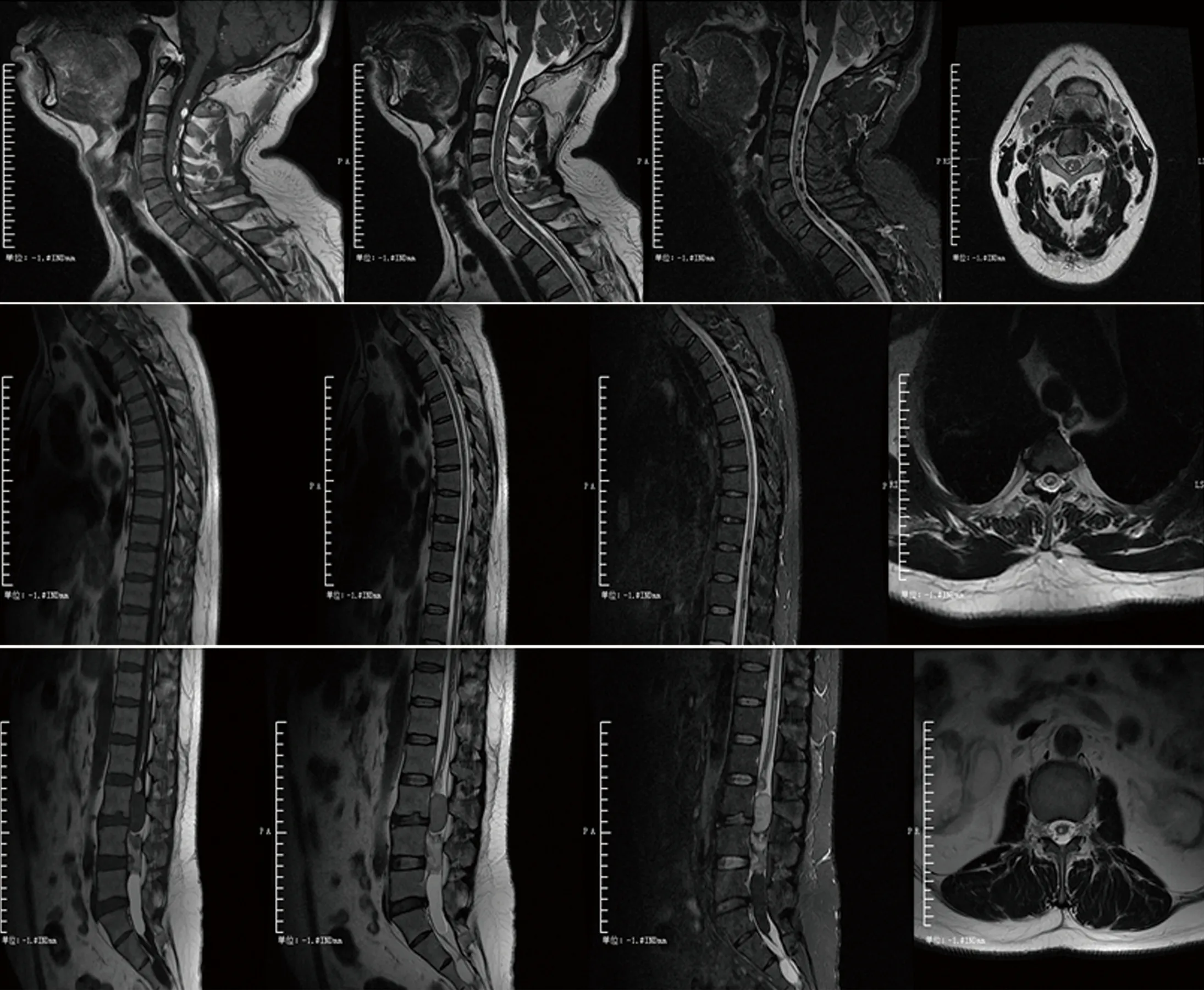
图2 颈髓、胸髓、腰髓内多发脂肪信号

图3 骶尾部脂肪瘤
2 讨 论
该患自幼起病,以皮下多处肿物伴有尿便障碍为主,成年后发现逐渐出现性功能障碍及肢体麻木感。对比患者2020年9月17日头部+脊椎MRI检查结果提示体内脂肪瘤无明显增多及增大,再次证实MSL诊断及其慢性进展病程的特点。该病例脂肪瘤主要分布于脑沟、脑池、脑室、外侧裂、脊髓后索、侧索、圆锥、马尾及皮下。其中L1-L2、S1-S2、T7-T12、S1-S3及S2-S4的脊髓侧角受累明显,故产生上述症状及相应的体征。目前关于MSL发病机制较为认可的是线粒体基因损伤及染色体异常学说,线粒体基因发生突变或损伤可使细胞色素氧化酶C、儿茶酚胺活性降低及腺苷酸环化酶合成障碍,造成脂肪代谢异常引起MSL。关于MSL基因变异方面,曾发现线粒体DNA中存在m.83 44A>G突变[3,4]。在伴有神经病变的MSL患者中也陆续发现线粒体基因MFN2 P基因的P.R 707w及c.2119G>T纯合子突变[5]。同时有权威机构研究表明,MSL患者脂肪细胞中miR-125a-3p和miR-483-5p异常增多,并可通过抑制RhoA/ROCK/ERKl/2通路促进脂肪堆积[6]。本次基因检测结果:线粒体基因检测了16569个位点,其中检测出突变基因ND2,ND2基因所处位置在线粒体基因组第5466位,突变形式是由A碱基变为了G碱基,引起的氨基酸变化为苏氨酸(T)变为了丙氨酸(A)。该基因检测的覆盖度为99%;正常人群携带率为0.00048;该患的基因位点突变频率为(3859-24)/3859,属于纯合突变。至今线粒体ND2基因突变在MSL临床研究上未有报导。棕色脂肪被认为是MSL的起源[7],因在脂肪瘤内发现了棕色脂肪组织特异性标志物(线粒体载体蛋白-分子解耦连蛋白I,UCP1,CD200),并且MSL发病的部位与棕色脂肪的位置大致相同。长期大量饮酒是MSL发病的一个诱发因素,酒精可导致线粒体中正常生理酶的代谢过程发生紊乱,造成线粒体代谢障碍、DNA氧化及导致早衰。过量的乙醇还会通过降低β-肾上腺受体的数量和活性进一步影响肾上腺素的脂解功能 从而诱发MSL[8,9]。目前也有相关研究证实仍有33%的MSL患者无饮酒史,表明饮酒为MSL的一个重要诱因,并非致病因素。
本次报导的MSL病例属于Enzi[10]分型中的Ⅰ型。Ⅰ型主要为男性患者,可有长期饮酒史,病变主要分布于颈项部、上背部、肩部、胸腹部及四肢近端,多数主要分布于颈部,外形似“河马颈”。据相关统计85%的MSL患者可有周围神经损伤,主要累及感觉和运动系统,其中自主神经亦可受累及。结合本次肌电图结果,考虑患者的双侧前角或前根受损,该病例感觉神经波幅处于正常低值,随着病情进展尚有累及周围神经的可能性。中枢神经系统受累少见,目前国内外仅报导10余例。其主要临床表现有小脑性共济失调、记忆力减退、听力障碍、精神症状、视神经萎缩、近端性肌病、锥体束征、认知功能下降和与年龄不匹配的脑萎缩等[11,12]。 MSL在MRI呈现T1呈短信号,T2呈中长信号,脂肪抑制后为T2低信号,如有纤维组织则可呈等信号。 增强下无明显强化,若存在索状纤维结构则可显示轻度强化。病理检查为MSL诊断的金标准,镜下病灶由无包膜的脂肪构成,其内可见脂肪细胞、纤维及血管组织及少量炎细胞浸润。目前认为该肿瘤性质介于良性肿瘤及恶行肿瘤之间,极少恶变,但已有可转化为髓内肉瘤及脂肪肉瘤的病例被报导[13]。实验室检查:主要包括线粒体基因检测,因相关报导称一些代谢性疾病常与MSL发病有关,故血脂、血糖、尿酸及甲功等检查也是必要的。MSL主要根据其临床特点诊断,同时需要与单纯性肥胖、库欣综合征、甲减、HIV药物使用导致的脂肪堆积及激素的使用、神经纤维瘤病、肌纤维营养不良、脂肪肉瘤、血管脂肪瘤、冬眠瘤、脂肪垫等疾病相鉴别[14,15]。
目前对MSL无病因方面的针对性治疗,一般治疗包括戒酒、低盐、低脂及建立健康的生活方式等。相关研究表明,戒酒可减少MSL的发生率及术后复发率,但不可治愈疾病。外科手术治疗是目前推荐的治疗方法[16],主要根据病灶的大小、累及周围组织情况及产生的并发症来合理使用。与脂肪切除术相比,抽脂术创伤小且更容易执行,然而,复发的风险是较高的,因为它很难完全消除脂肪瘤。目前使用溶脂药物治疗MSL的病例也有报导,有学者证实注射磷脂酰胆碱存在一定的有效性[17 ],但仅起到暂时性的改善作用。综上,为进一步防治MSL,对MSL的病因及临床特性的研究将是临床及科研工作的重点。
