甲状腺癌中m6A甲基化调控因子的表达及其预后价值
2021-09-09王文龙沈聪孙博韬白宁李新营陈嘉欣彭婀敏
王文龙,沈聪,孙博韬,白宁,李新营,陈嘉欣,彭婀敏
(中南大学湘雅医院1.甲状腺外科3.国际医疗部,湖南长沙410008;2.中南大学湘雅医学院,湖南长沙410013)
过去几十年,甲状腺癌的发病率在全世界范围内迅速上升[1-2]。据国家癌症中心数据显示,甲状腺癌已跻升至女性恶性肿瘤的第3 位,且发病出现年轻化趋势,已成为我国30 岁以下女性增长最快的恶性肿瘤[3]。甲状腺乳头状癌是最常见的病理亚型,占85%以上[4-5]。手术是其主要治疗方式,规范化手术治疗后10年生存率达90%以上[6],但仍有1%~9%的患者在初次就诊时就已出现远处转移,严重影响了患者预后[7]。因此,阐述甲状腺癌的发病机制对改善预后至关重要。
m6A 甲基化修饰是指RNA 的腺苷酸的第6 位N处发生动态可逆的甲基化过程,在调控mRNA 的转录、翻译、稳定、剪切、成熟等发挥了重要的生物学作用,其功能主要由甲基转移酶(writer)、去甲基化酶(eraser)和识别蛋白(reader)决定[8-10]。越来越多的证据表明m6A 甲基化修饰在肿瘤的发生发展扮演了重要的角色[9,11-13]。METTL3 通过YTHDF2 依赖的途径上调抑癌基因SOCS2 的m6A 甲基化水平,从而加速SOCS2 mRNA 的降解。METTL3 过表达促进肝癌细胞的增殖和迁移,相反,敲低METTL3 显著抑制肿瘤生长和转移[14]。研究[15]揭示了IGF2BP3 在结直肠癌中高表达,下调IGF2BP3 可以通过VEGF 和CCND1 的m6A 修饰来抑制血管生成和DNA 复制。以上研究证实m6A 甲基化调控因子高度参与癌症发生发展,具有良好的潜在预后价值。然而,目前对于m6A 甲基化调控因子在甲状腺癌致病调控机制尚不清楚。
本研究利用癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库中甲状腺癌RNA-sep数据,系统性分析了20 个m6A 甲基化调控因子在甲状腺癌中的表达水平,再利用聚类分析和Lasso Cox 回归分析筛选出预后相关基因及构建风险预测模型并进行危险分层,为甲状腺癌患者的临床诊疗和预后改善提供了重要的理论参考依据。
1 资料与方法
1.1 数据的获取与处理
从TCGA 数据库(https://portal.gdc.cancer.gov)下载了包含510 例甲状腺癌组织和58 例正常组织样本的RNA-seq 数据集及相应临床数据。临床病理信息包括性别、年龄、肿瘤学多灶性、TNM 分期、淋巴结转移、组织学类型、肿瘤位置和生存时间等,剔除RNA 序列以及临床病理数据不全的样本,共492 例癌组织和58 例正常组织样本纳入最终分析。所有患者的病理分期按照美国癌症联合委员会(AJCC)2018年1月颁布了第8 版甲状腺癌TNM分期。根据之前m6A 甲基化在肿瘤中的研究[11,16],共筛选20 个m6A 甲基化调控因子,包括甲基转移酶(WTAP、METTL3、METTL14、METTL16、KIAA1429、ZC3H13 和 RBM15)、识 别 蛋 白(HNRNPA2B1、IGF2BP1/2/3、YTHDC1/2、FMR1、LRPPRC 和YTHDF1/2/3)和去甲基化酶(ALKBH5和FTO)。应用R 语言的“Limma”软件包分析上述基因在癌与正常样本的差异表达。
1.2 聚类分析
为了从功能上阐明甲状腺癌中m6A 调控因子的生物学特性,使用“consensusclusterplus”软件包对甲状腺癌患者进行一致性聚类分析并将患者分成cluster 1 和cluster 2 组。主成分分析(PCA)检测基于m6A 调控因子表达是否能将两组患者明显区分,并探讨不同的临床病理因素和总体生存率在两组聚类中是否存在差异。
1.3 风险预测模型的建立与评估
为了探讨m6A 调控因子在甲状腺癌预后的作用,通过“glmnet”软件包进行Lasso Cox 回归分析筛选预后相关的基因,并计算基因的相关回归系数,根据回归系数加权建立甲状腺癌预后风险评估公式,risk score=Coei*Expi,Coei表示回归系数,Expi表示m6A 调控因子的表达量。据此公式计算每个患者的风险评分,以风险评分的中位数将患者分为高危组和低危组,比较两组总体生存率是否存在差异,ROC 曲线下面积(AUC)评估模型的预测能力。
1.4 统计学处理
采用R 3.6.0 软件进行统计学分析,Wilcoxon 秩和检验分析20 个m6A 甲基化调控因子在肿瘤和正常组织的差异表达。计量资料用t检验,计数资料用χ2检验。采用Spearman 相关分析基因间的相关性,0.1≤|r|≤1.0 时定义为存在相关;生存曲线采用Kaplan-Meier 法和Log-rank 检验,P<0.05 为差异有统计学意义。
2 结果
2.1 一般资料及m6A 甲基化调控因子的基因表达与相关性
本研究共纳入492 例甲状腺癌患者,平均年龄47.29 岁,男女比列1∶2.76;临床病理TNM 分期:I 期276 例,II 期52 例;III 期109 例,IV 期55 例;T 分期:T1期135例,T2期162例,T3期172例,T4期23例;44.9%的患者出现颈部淋巴结转移;9 例患者出现远处转移;228 例肿瘤呈现出多灶性,占46.3%;肿瘤位于峡部、左侧、右侧、双侧分别为22、172、214,84 例(表1)。
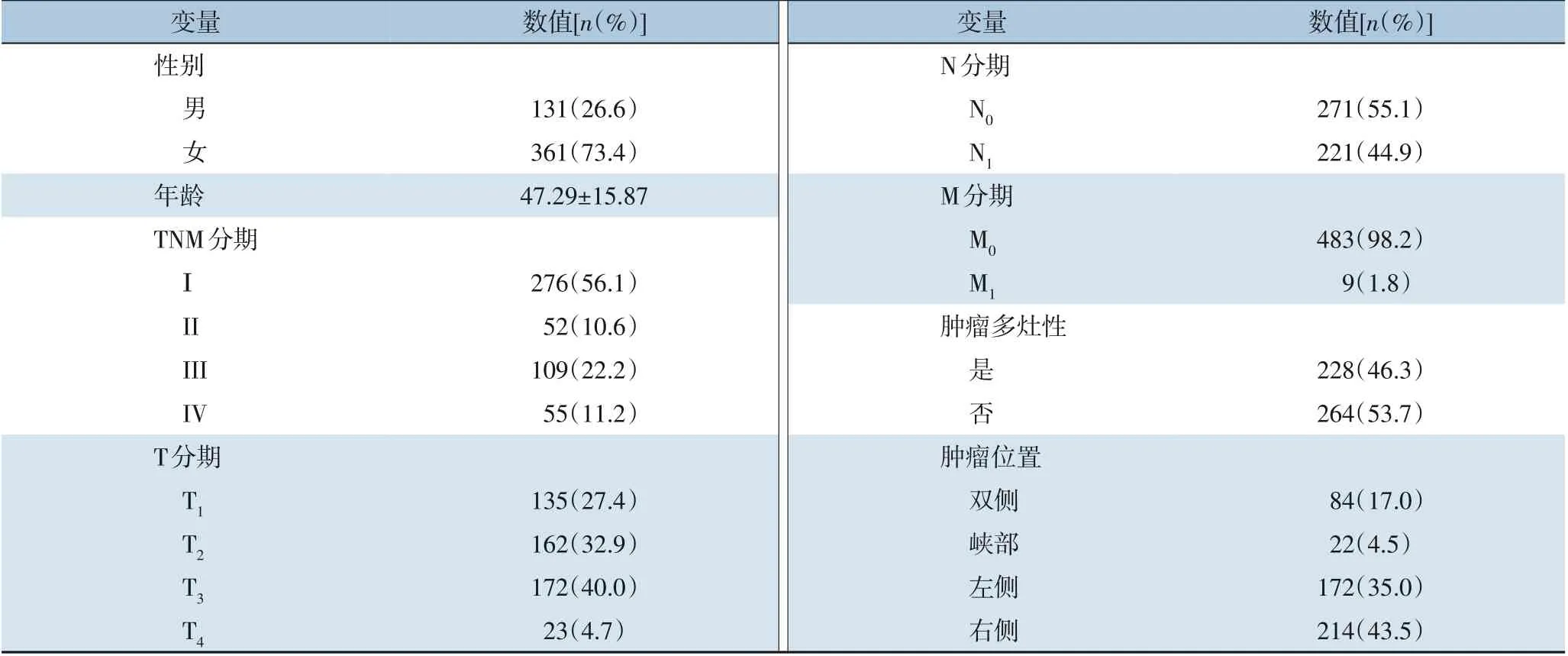
表1 492例甲状腺癌患者基本临床病理资料Table 1 The clinicopathologic information of the 492 patients with thyroid cancer
19 个m6A 甲基化调控因子在甲状腺癌和正常组织表达具有统计学差异(P<0.05),其中HNRNPC(P<0.001)、IGF2BP2(P<0.001)、FMR1(P<0.05)在甲状腺癌组织中明显高表达。相反,ALKBH5(P<0.001)、FTO(P<0.001)、METTL3(P<0.001)、METTL14(P<0.001)、METTL16(P<0.05)、WTAP(P<0.001)、YTHDF1(P<0.001)、YTHDF3(P<0.001)、YTHDC2(P<0.001)、YTHDC1(P<0.001)、ZC3H13(P<0.001)、HNRNPA2B1(P<0.001)、RBM15(P<0.001)、IGF2BP1(P<0.001)、IGF2BP3(P<0.001)和LRPPRC(P<0.001)在甲状腺癌标本中表达明显下调(图1A)。此外,大部分m6A 甲基化调控因子都显示正相关,其中,METTL14 与YTHDC1、YTHDF3与ZC3H13 呈现最强正相关(r=0.73);而ALKBH5 与IGF2BP2呈明显负相关(r=-0.33)(图1B)。为了进一步了解19 个m6A 甲基化调控因子相互关系,PPI 构建了网络相互作用网(图1C)。
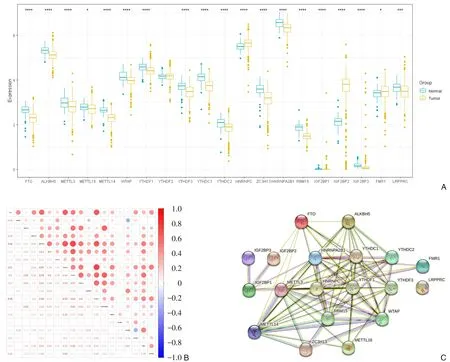
图1 m6A甲基化调控因子的基因表达及相关性A:20个m6A甲基化调控因子在甲状腺癌和正常组织表达水平;B:Spearman相关分析m6A甲基化调控因子相关性;C:PPI网络评估m6A甲基化调控因子相互作用关系Figure 1 Gene expression and correlations of the m6A methylation regulatorsA:The mRNA levels of the 20 m6A regulators in thyroid cancer; B:Spearman correlation analysis of co-expressions among the m6A regulators; C:PPI network of the m6A regulators
2.2 一致性聚类分析m6A甲基化调控因子
为了确定最佳的聚类数,采用一致性聚类分析对492 例甲状腺癌患者进行聚类分组,共识矩阵k 值为2 时,甲状腺癌样本之间不存在交叉,聚类稳定性好(图2A)。因此,492 例甲状腺癌患者被分成cluster 1 和cluster 2 组,主成分分析可以将两组患者明显区分(图2B)。聚类热图示,cluster 1组颈淋巴结转移发生率明显高于cluster 2(P<0.01),而TNM 分期、T 分期、M 分期、肿瘤多灶性、肿瘤位置、年龄、性别在两组之间无差异(均P>0.05)(图2C)。生存分析示,cluster 1 生存时间低于cluster 2(P=0.048)(图2D)。

图2 一致性聚类分析m6A甲基化调控因子A:k值为2时,甲状腺癌样本之间不存在交叉,聚类稳定性好;B:主成分分析区分cluster 1和cluster 2;C:不同的聚类与临床病理的关系;D:总体生存率在两组聚类的情况Figure 2 Consensus clustering analysis of the m6A RNA methylation regulatorsA:The most appropriate selection with clustering stability when k=2 used; B:Principal component analysis to distinguish cluster 1 and cluster 2; C:Heatmap and clinicopathologic features of the two clusters;D:The overall survival of cluster 1 and cluster 2
2.3 m6A甲基化调控因子构建风险预后模型及检验
Lasso Cox 回归分析筛选4 个基因(IGF2BP2、RBM15、YTHDF1、YTHDF3),风险评分=(-0.129×IGF2BP2)+(0.308×RBM15)+(0.331×YTHDF1)+(0.813×YTHDF3)(图3A),根据回归系数计算每个患者的风险评分,以风险评分的中位数将患者分为高危组和低危组,风险评分和生存状态分布如图3B 所示,高风险患者中死亡人数明显增加。相比低风险组患者,高风险患者生存期明显缩短(P=0.007)(图3C),ROC 曲线下面积示该模型可预测甲状腺癌患者的预后(AUC=0.731)(图3D)。
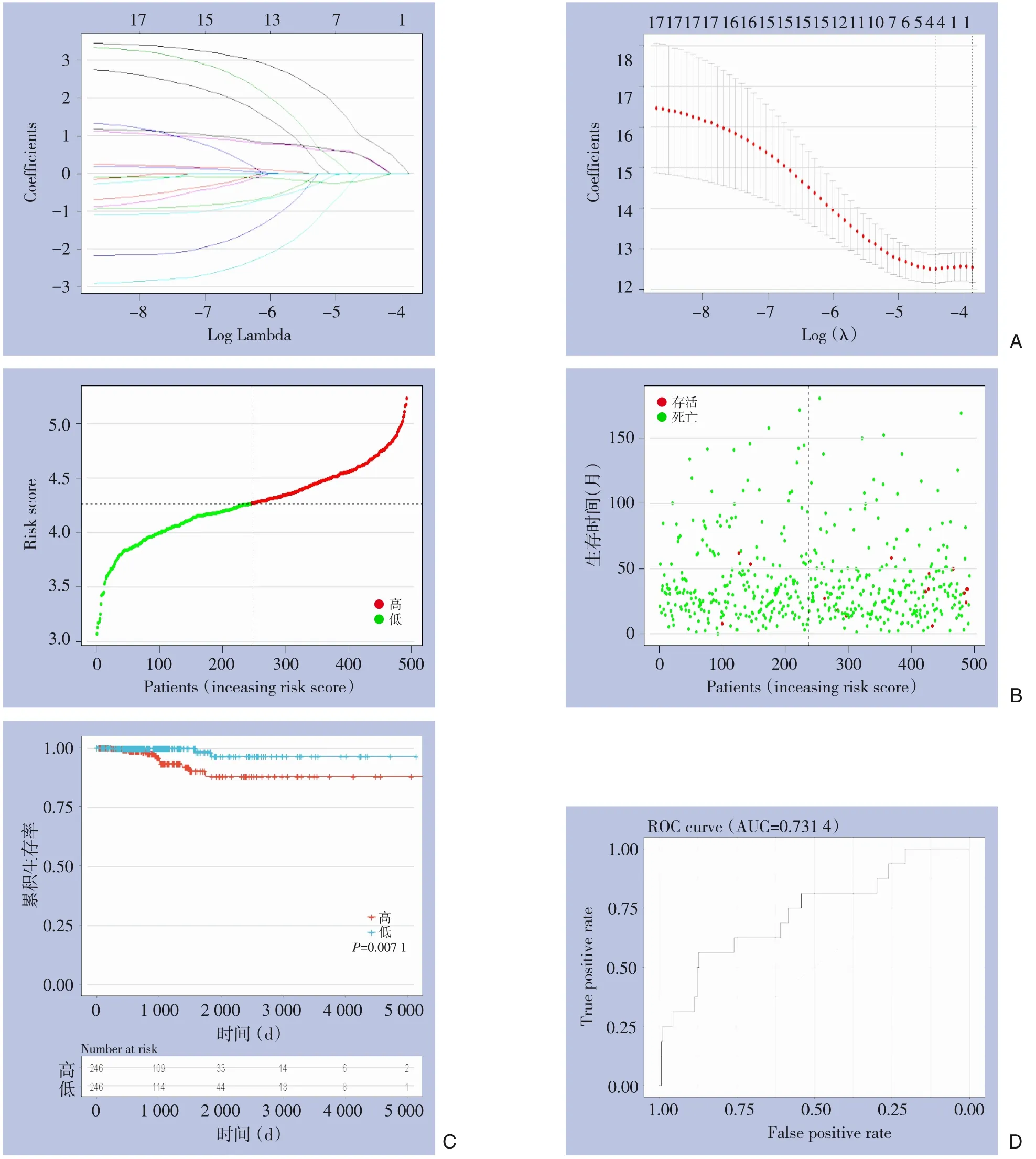
图3 m6A甲基化调控因子构建风险预后模型及检验A:Lasso Cox回归模型的m6A基因及相关系数;B:风险评分和生存状态分布;C:高、低危组的生存曲线;D:ROC曲线评估模型的预测效能Figure 3 Construction od prognostic risk model and validationA:Coefficients calculated by Lasso Cox regression analysis;B:Risk score and survival status distribution;C:Kaplan-Meier analysis of overall survival in high-risk and low-risk groups;D:Assessment of the prediction accuracy by ROC curve
3 讨论
甲状腺癌是一种常见的内分泌恶性肿瘤,发病率在世界范围内呈快速上升趋势,绝大部分甲状腺癌预后良好[17-18]。然而一些分化较差的甲状腺癌和高危型甲状腺乳头状癌具有高度侵袭性,易出现局部侵犯和远处转移,且尚无特效治疗方法[19-20]。因此阐述甲状腺癌发生和演变进展的分子机制对疾病的早期干预和预后预测具有重要的临床应用价值。
m6A 修饰是近年来的研究热点,其作用主要参与转录后修饰,同时也参与了DNA 修复、细胞分化、细胞重编程、细胞应激等生命活动[21-22]。m6A 甲基化在不同的肿瘤中均有报道,在甲状腺癌中m6A 相关基因的作用仍不清楚,Ye 等[23]发现IGF2BP2 在甲状腺癌中高表达,lncRNA MALAT1 通过竞争性结合miR-204,通过m6A 修饰识别,上调IGF2BP2,增强MYC 表达,从而刺激甲状腺癌细胞增殖、迁移和侵袭,同时减弱肿瘤生长和细胞凋亡。然而该研究样本量有限无法验证lncRNA MALAT1/mir-204/IGF2BP2/m6A-MYC 轴在甲状腺癌进展机制。Wang 等[24]报道METTL3 作为致癌基因通过上调TCF1 的m6A 甲基化水平促进甲状腺癌的增值和侵袭。然而He 等[25]研究揭示METTL3 作为抑癌基因结合YTHDF2 通过c-Rel 和RelA 失活NF-κB 通路改变肿瘤相关的中性粒细胞浸润来调节肿瘤生长。同样,Hou 等[26]通过生物信息学证实METTL3 在甲状腺癌组织中明显低表达。上述研究证实m6A 相关基因表达水平和功能机制在甲状腺癌中仍需进一步阐述。本研究初步验证了METTL3 在正常组织中表达量明显高于甲状腺癌组织(P<0.001),提示METTL3 可能是一种抑癌基因,可改善甲状腺癌患者的预后,这也进一步验证了He 等[25-26]的研究结果。
为了从功能上阐明甲状腺癌中m6A 调控因子的生物学特性,本研究对492 例甲状腺癌患者进行一致性聚类分析后并分成cluster 1 和cluster 2,cluster 1 生存期低于cluster 2(P<0.05),相反,颈淋巴结转移发生率明显高于cluster 2(P<0.01)。暗示m6A 调控因子能预测颈淋巴结转移并影响甲状腺癌患者预后,具有非常重要的临床实际意义。聚类分析在肝癌[27]、肾癌[16]、乳腺癌[11]、胃癌[28]等恶性肿瘤预后分析中均起到较好的作用。
此外,为了探讨m6A 调控因子在甲状腺癌预后的作用,应用Lasso Cox 回归分析筛选的4 个基因(IGF2BP2、RBM15、YTHDF1、YTHDF3)构建风险评估模型,相比IGF2BP2,其余3 个基因在甲状腺癌的研究中甚少,RBM15 不具有甲基转移酶的催化功能,但它可以结合METTL3 和WTAP,并将这两个蛋白引导到特定的RNA 位点进行m6A 修饰[8,29];而YTHDF1 和YTHDF3 均属于YTH 域家族,YTHDF1 通过与起始因子相互作用增强mRNA 翻译和蛋白质合成,YTHDF3 分别通过与YTHDF1 相互作用增强RNA 翻译,与YTHDF2 结合促进RNA 降解[9,30]。相比低风险组患者,高风险患者生存期明显缩短(P=0.007),ROC 曲线示该模型可以预测甲状腺癌患者的预后(AUC=0.731)。相比TNM 分期和MACIS 预后评估系统,该模型基于RNA 测序数据,更符合当今的精准医学理念。
同时,本研究也存在一些缺陷,首先,该研究基于生物信息学构建的预后预测模型,无外部的实验数据验证,临床实际应用价值有限。其次,该研究只是初步探讨了m6A 调控因子在甲状腺癌预后作用,不能完整的阐述甲状腺癌发生和演变的分子机制。最后,本研究基于492 例甲状腺癌患者,样本量不大,后续需要进行更大的独立队列研究来验证。
综上,本研究基于TCGA 数据库分析甲状腺癌m6A 调控因子表达水平,通过Lasso Cox 回归分析筛选的4 个基因(IGF2BP2、RBM15、YTHDF1、YTHDF3)构建风险评估模型,AUC 提示该模型具有良好的预测效能,将为甲状腺癌临床诊治提供潜在的理论依据。
