甲基苯丙胺、可卡因和吗啡成瘾对糖代谢的影响及分子机制研究进展
2021-09-07温先槟
温先槟,白 洁
(昆明理工大学医学院,云南 昆明 650500)
药物成瘾是一种慢性复发性脑疾病,主要表现为强迫性地连续或定期用药行为[1],一旦停药则出现精神和躯体不适。药物成瘾者为得到药物不择手段,给社会带来安全隐患,使自己和家人背上沉重的经济负担。
成瘾药物主要包括甲基苯丙胺(又称去氧麻黄碱,methamphetamine,METH)、吗啡、可卡因、氯胺酮和大麻素等,而其中METH、吗啡和可卡因滥用一直是威胁全球临床和公共卫生的严重问题[2]。METH和可卡因是中枢神经系统(central nervous system,CNS)兴奋剂,METH作用时间较长,可卡因作用时间较短。吗啡对大脑皮质痛觉区有抑制作用,是一种常用的镇痛药物。研究表明,多巴胺(dopamine,DA)能、谷氨酸能和γ-氨基丁酸(γaminobutyric acid,GABA)能神经系统在药物成瘾中起重要调节作用[3-5],但调节机制尚不清楚。最近发现,DA、谷氨酸和GABA与糖代谢途径密切相关。DA可抑制葡萄糖(glucose,Glu)诱导的胰岛素分泌[6]。星形胶质细胞介导的谷氨酸循环和GABA代谢的能量需求主要来自于糖酵解、Glu代谢和乳酸产生[7]。研究发现,胰岛素对中脑腹侧被盖区(ventral tegmental area,VTA)的 GABA 受体和α-氨基-3-羟基-5-甲基-4-异唑丙酸受体(alphaamino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor,AMPAR)等具有调节作用[8-9]。胰岛素可能通过调节DA受体、GABA受体和AMPAR影响DA能、谷氨酸能和GABA能神经系统的功能,改善药物成瘾引起的糖代谢紊乱。
1 糖代谢与胰岛素、PI3K/Akt和AMPK信号通路
糖代谢是指Glu和糖原等糖类物质在体内生物转化的生化过程,而糖代谢研究主要集中在Glu的代谢。Glu是维持机体内所有器官组织正常运行的能量物质。因此,保持糖代谢动态平衡至关重要。
胰岛素和胰高血糖素是调节糖代谢动态平衡的重要激素。胰岛素由胰岛β细胞分泌,主要通过诱导脂肪组织形成、骨骼肌糖吸收和肝糖原合成,及抑制肝糖原分解和糖异生等降低血糖水平。胰高血糖素由胰岛α细胞分泌,为胰岛素的主要拮抗激素,其主要靶器官为肝、肾和心肌细胞等。胰高血糖素促进肝糖原分解,抑制肝糖原合成,促进糖异生和分解,并能促进脂肪分解,导致血糖升高(图1)。
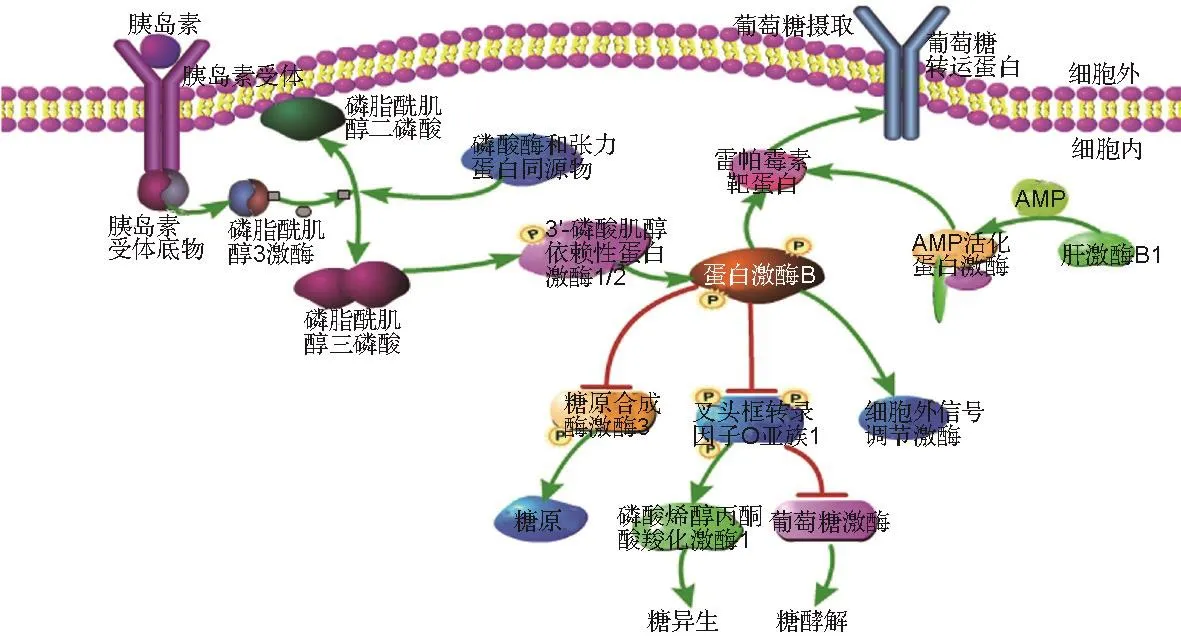
图1 胰岛素调节糖代谢的信号通路.P:磷酸化;→:促进作用;┤:抑制作用.
1.1 胰岛素信号通路
胰岛素主要通过磷脂酰肌醇3激酶(phospha⁃tidylinositol 3-kinase,PI3K)/蛋白激酶 B(protein kinase B,Akt)和AMP活化蛋白激酶(AMP activated protein kinase,AMPK)等信号通路在肝、肌肉和神经细胞中发挥多种作用[10-12]。
胰岛素通过胰岛素受体(insulin receptor,INSR)发挥降血糖作用。INSR是酪氨酸激酶受体的一种,是由2个α亚基和2个β亚基组成的四聚体。INSR底物(INSR substrates,IRS)可被具有激酶活性的β亚基磷酸化,从而结合具有SH2结构域的蛋白[8-9]。活化的IRS能激活下游的PI3K/Akt信号通路,以及哺乳动物雷帕霉素靶蛋白(mammalian target of Rapamycin,mTOR)信号通路,从而调节Glu代谢[13]。
在CNS,INSR主要分布于内嗅区、下丘脑、大脑皮质、小脑和海马等脑区的神经元和星形胶质细胞中[14]。INSR缺失影响大脑内神经元的数量,如DA能神经元和GABA能神经元,而GABA能神经元对胰岛素调节外周糖代谢起重要作用[15]。胰岛素通过PI3K和丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPK)通路调节线粒体的功能。下丘脑作为调节食欲的关键核团,对胰岛素水平的变化作出应答,如在给大鼠侧脑室注射胰岛素后,会导致食物摄入量的减少和饱腹感激素的增加[16-17]。而选择性降低下丘脑INSR表达,将造成大鼠摄食过度[16]。鼻腔内胰岛素给药可提高人脑脊液中的胰岛素水平,增强外周胰岛素敏感性,抑制内源性Glu产生[15]。当在神经元能量需求超过Glu供应时,大脑中的星形胶质细胞将其储存的糖原转化为Glu,通过葡萄糖转运体1(glucose transporter 1,GLUT1)转运至胞外,从而为神经元活动提供Glu[18]。
1.2 PI3K/Akt信号通路
PI3K/Akt通过GLUT4和INSR维持Glu的稳态[19-20]。PI3K能使磷脂酰肌醇二磷酸〔phosphati⁃dylinositol(4,5)-bisphosphate,PIP2〕被特异性地转化为PIP3,而PIP3通过与Akt的PH结构域结合,使其活化[21];活化的Akt通过调节糖原合酶激酶3(glycogen synthase kinase 3,GSK3)和细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)等相关下游分子增加糖原的生成。同时,磷酸化转录因子叉头框蛋白O1使其失活,促进糖异生,抑制糖酵解,降低血糖,从而维持Glu的稳态。此外,CNS的IRS/Akt信号通路激活,通过ERK/环磷酸腺苷反应元件结合蛋白(cAMP-response element binding protein,CREB)/脑源性神经营养因子(brainderived neurotrophic factor,BDNF)信号转导途径,改善胰岛素抵抗和认知功能障碍,在SH-SY5Y细胞中能逆转高葡萄糖和氨基葡萄糖诱导的胰岛素抵抗[22]。综上,PI3K/Akt信号通路不仅是糖代谢的重要信号通路之一,还介导多种生长因子作用。
1.3 AMPK信号通路
AMPK即AMP依赖的蛋白激酶,是细胞能量代谢调节的关键分子。AMPK是能量传感器和调节剂,可促进组织中ATP的产生,并抑制ATP的消耗途径。AMPK是异源三聚体复合体,由催化性α亚单位和调节性β和γ亚单位组成。AMP结合到γ亚单位后,导致复合体变构激活。AMPK是肝激酶B1(liver kinase B1,LKB1)的直接底物,LKB1通过促进α亚基上苏氨酸172位点的磷酸化增强AMPK的磷酸化水平,从而使AMPK激活[23]。AMPK的激活促进糖酵解,产生能量,增加了GLUT4的转录和易位,从而促进胰岛素将血液中Glu摄入组织细胞内。综上,LKB1-AMPK通路能通过调节肝Glu产生和糖异生,维持Glu稳态[24]。
2 成瘾药物与糖代谢异常
成瘾性药物作用于CNS,导致神经递质释放异常,神经递质与糖代谢密切相关。而CNS的胰岛素敏感性降低、糖代谢紊乱均可导致大脑结构与功能改变,这些改变与成瘾药物所致成瘾行为有关。
大脑默认模式网络(default mode network,DMN)是指大脑在无任务的静息状态下,仍进行着某些功能活动的脑区所构成的网络,与大脑对内外环境的监测和情景记忆的提取等功能相关。最近研究发现,无论是急性还是慢性给予成瘾药物,均会导致DA能、谷氨酸能和GABA能神经系统适应性改变,造成神经系统DMN损伤,导致成瘾[25]。METH和可卡因通过与多巴胺转运体(dopamine transportor,DAT)和囊泡单胺转运体结合,干扰DA再摄取和包装系统,使DA释放水平上升,从而产生奖赏和依赖效应[19-20]。
胰岛素的代谢与DA等神经递质有关[15,26],DA可抑制胰岛β细胞分泌胰岛素[27]。胰岛素信号通路与成瘾相关信号通路在CNS相互联系,如吗啡可导致VTA区胰岛素生长因子1(insulin growth factor-1,IGF-1)和IRS表达水平下降,胰岛素通过调节阿片肽、GABA和内源性大麻素等神经递质抑制药物成瘾造成的糖代谢异常[8]。重要的是,胰岛素还对突触可塑性具有调节作用[28],而突触可塑性在药物成瘾中起关键作用[9]。
2.1 METH与糖代谢
METH呈透明结晶体状,纯品形似冰,俗称冰毒,是新型毒品的一种[29]。METH滥用可导致血糖水平降低[30]。研究发现,METH通过抑制胰岛细胞中p-Akt,GSK-3α/β和p-ERK水平,影响胰岛素信号通路,导致糖代谢异常[31]。给予SH-SY5Y细胞不同浓度的METH孵育12 h后,Akt-Ser473磷酸化水平明显降低,GSK-3α/β磷酸化水平也相应降低,GSK-3α/β被激活,抑制胰岛素信号转导,从而导致糖代谢异常[32]。研究发现,给大鼠ip给予METH(1.5~5.0 mg·kg-1,不断增加METH剂量,持续14 d),会降低大鼠心肌细胞中p-Akt,p-GSK3和p-ERK的水平,引起糖代谢紊乱,进一步造成大鼠心肌损伤和凋亡[33]。胰岛素可逆转METH引起的条件性位置偏爱(con⁃ditioned place preference,CPP),抑制METH诱导的大鼠奖赏行为;而胰岛素水平降低将增强METH诱导的奖赏效应,给予胰岛素后,METH奖赏效应则被抑制[34]。大鼠连续7 d,每天双侧鼻腔内给予胰岛素0.5 U(2.5 μL),激活胰岛素信号转导途径,可减轻METH引起的认知障碍,从而改善METH成瘾[35]。综上所述,METH通过抑制胰岛素信号转导途径,导致糖代谢异常,而胰岛素可以改善METH引起的糖代谢紊乱,进而治疗METH成瘾。
2.2 可卡因与糖代谢
可卡因是一种精神兴奋类的成瘾性药物,主要作用于伏隔核(nucleus accumbens,NAc)的DA神经元末梢,与突触囊泡中的DAT结合,降低DAT活性,抑制突触前膜对突触间隙DA的再摄取,间接增加NAc区DA水平;DA水平增高,则可导致D2样受体调节的钾离子通道的激活,Ca2+内流减少,导致胰岛β细胞内Ca2+浓度降低,抑制胰岛素分泌,而D2样受体拮抗剂可逆转这一作用[27,36]。也有研究发现,DA浓度增加可激活D1受体,上调ERK,Akt和CREB的磷酸化水平[37],抑制Glu刺激的胰岛素分泌。由此可见,可卡因导致的DA升高不仅与奖赏效应有关,还与糖代谢密切相关。大鼠VTA核团内微量注射胰岛素后,抑制可卡因所致的NAc区DA的释放和大鼠运动活性的改变,而在VTA核团内微量注射INSR拮抗剂S961后,则阻断胰岛素这一作用[38]。同样,大鼠经鼻腔内给予胰岛素,则降低可卡因所致的奖赏效应和运动活性[38]。胰高血糖素样肽1(glucagon-like peptide 1,GLP-1)是一种小肠和孤束核分泌的脑肠肽,促进胰岛素的分泌。研究发现,GLP-1受体激动剂唾液素4可抑制可卡因的奖赏效应,提示GLP-1受体可能成为可卡因成瘾的治疗靶点[39]。有研究发现,可卡因给药后,猕猴大脑糖利用增加[40]。产前暴露可卡因等精神兴奋类物质可导致胎儿糖代谢紊乱[41]。这些研究说明糖代谢紊乱与可卡因成瘾密切相关。因此,胰岛素可用于治疗可卡因成瘾。
2.3 吗啡与糖代谢
吗啡作为镇痛类阿片药物,具有镇痛和镇咳作用,长时间使用能导致成瘾。吗啡通过结合细胞表面的阿片受体,激活G蛋白偶联受体,导致腺苷酸环化酶活性的降低,钾离子通道激活和钙电导下降;吗啡还具有调节磷脂酶C和MAPK信号通路的作用[42]。大鼠吗啡自给药模型中,杏仁核内INSR的表达急剧减少,而戒断后INSR的表达逐渐恢复[43]。利拉利汀(linagliptin)是二肽基肽酶Ⅳ的抑制剂,可抑制GLP-1降解,降低血糖。研究发现,利拉利汀可抑制吗啡CPP的表达,促进吗啡CPP的消退,还可抑制吗啡CPP点燃[44]。此外,有研究发现,二甲双胍通过激活AMPK,抑制mTOR,P38和MAPK的活性,改善吗啡诱导的神经炎症和镇痛耐受[45]。
研究报道,雌性大鼠孕前阿片类药物暴露增加了其子代代谢综合征/2型糖尿病的患病风险[46];而在雄性大鼠静脉注射阿片类药物后,则引起大脑缺氧和高血糖症[47]。用正电子发射断层扫描成像技术发现,青年(4周龄)大鼠吗啡给药后,其大脑糖代谢显著变化,其中胼胝体和右扣带运动皮质区Glu代谢增加,而右腹侧苍白球Glu代谢减少;在胼胝体、右扣带运动皮质区和右腹侧苍白球中,GLUT3、D2受体和μ阿片的表达与Glu代谢呈正相关[48]。综上所述,吗啡可通过AMPK信号通路和胰岛素信号通路影响CNS糖代谢,导致糖代谢异常;而恢复糖代谢平衡,可治疗吗啡成瘾。
3 结语
药物成瘾严重危害人体健康,关系到家庭幸福以及社会的和谐稳定,目前缺乏有效安全的治疗措施。成瘾药物导致DA系统、谷氨酸系统和GABA系统糖代谢紊乱,也改变了糖代谢稳态。METH、可卡因和吗啡等成瘾药物通过胰岛素、PI3K/Akt和AMPK信号通路抑制GLP-1和GLUT3表达,导致糖代谢紊乱。然而,成瘾药物通过调节糖代谢途径导致CNS糖代谢紊乱的机制尚不清楚,需进一步深入研究。
