假性甲状旁腺功能减退症1例
2021-09-06杨梦录王翠翠孙维梅李建厂
杨梦录 王翠翠 孙维梅 李建厂
滨州医学院附属医院儿科 山东 滨州 256603
假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是一种具有甲状旁腺功能减退症的症状和特征性体征的遗传性疾病。自腺体至靶组织细胞之间任何环节的缺陷均可以引起甲状旁腺功能减退,是一种少见的家族性疾病,典型患者有独特的骨骼缺陷和发育缺陷。主要表现为Albright遗传性骨营养不良症(AHO),即身材矮小、体胖、圆脸、颈短、盾状胸;短指、畸形趾;桡骨弯曲,软组织钙化或骨化较多见。患者临床表现为低钙性搐搦,实验室异常为血甲状旁腺素(parathyroid hormone,PTH)抵抗,即低血钙、高血磷、血PTH升高。本文对我院儿科收治的1例PHP患儿的临床特点及基因突变进行分析。
1 临床资料
患儿,女,8岁,因“反复抽搐发作5年,发热3 d”于2019年8月6日入院。
患儿约5年前无明显诱因出现抽搐,表现为意识丧失、四肢强直-阵挛、颈后仰、双眼凝视、口吐泡沫、口周发绀,持续时间2~3 min。于2014年7月14日至16日因“反复抽搐9 h”至我院住院治疗,查电解质示血钙为1.40 mmol/L(2.1~2.9 mmol/L),立即复查血钙为1.27 mmol/L(2.1~2.9 mmol/L),1,25-双羟维生素D为26.41 ng/mL(20~32 ng/mL),给予葡萄糖酸钙静点治疗,后因“吸气性呼吸困难、意识不清、烦躁不安”转至重症医学科,给予吸氧、抗感染、补充钙剂、止痉等治疗,患儿情况好转,复查血钙为1.63 mmol/L(2.1~2.9 mmol/L),家属拒绝继续治疗、完善相关检查,自动出院。出院后仍有间断抽搐发作,表现同前,每次抽搐时间2~5 min不等,一直未再行其他诊治。
近3 d出现发热,抽搐较前发作频繁,约5次/日,表现同前。2 d前出现咳嗽。既往无重大疾病史,无食物、药物过敏史。患儿系第2胎第1产(G2P1),足月剖宫产娩出,出生时有窒息缺氧史。患儿3个月抬头,12个月会坐,2.5岁说话,7岁会走,蹒跚步态,左脚跟不能着地,只能走平路。现未上学,智力水平明显低于同龄儿,身高增长也落后于同龄儿。父母体健,非近亲结婚,患儿外祖父及曾外祖母有抽搐病史,具体不详。入院体检,身高94.5 cm,标准差比值法(standard deviation score,SDS)值-6.30,体质量22.5 kg,体质量指数(body mass index,BMI):25.16,发育落后,表情淡漠。牙釉质发育不良,牙齿排列不齐。双肺呼吸音粗,未闻及干、湿性啰音。心律齐,心音低钝,未闻及杂音。手指粗短,双手第3、4指不能并拢,见图1。足趾粗短,见图2。肢体活动受限,两肘关节有变形,均不能伸直(最大角度约为150°),“O”型腿。
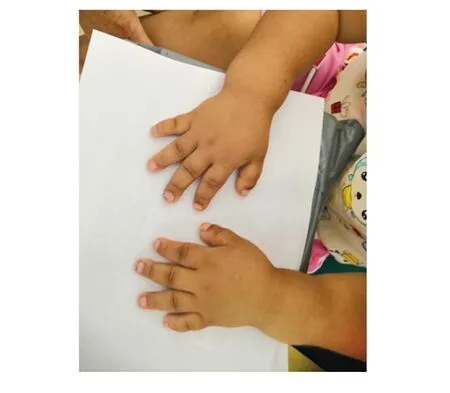
图1 患儿双手照片
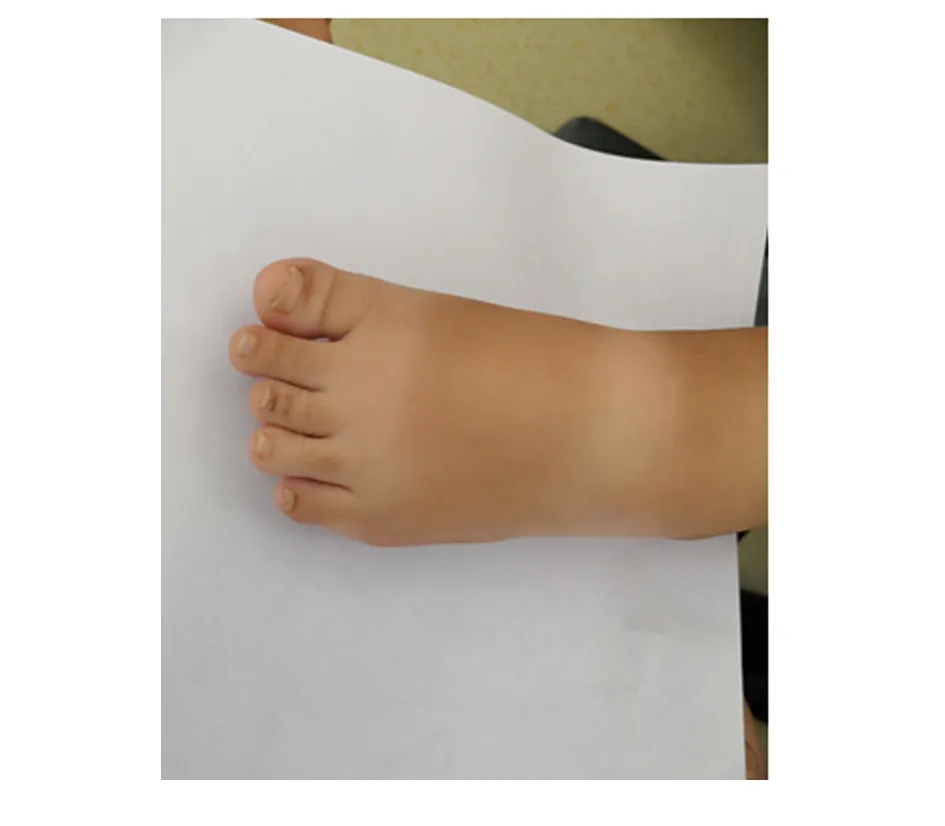
图2 患儿左足照片
辅助检查:血常规、大小便常规、肝功及肾功基本正常;肌酸激酶为1 029.2 U/L(25~200 U/L),肌酸激酶同工酶为27.5 U/L(≤19 U/L);血钙为1.44 mmol/L(2.2~2.7 mmol/L),血磷为2.55 mmol/L(0.85~1.51 mmol/L),血镁、血钾、血钠正常;性激素七项基本正常;25-羟维生素D为112.20 ng/mL(30~100 ng/mL);促甲状腺激素(TSH)为6.720 mIU/L(0.28~4.3 mIU/L),三碘甲状腺原氨酸(T3)为3.44 pmol/L(3.8~8.6 pmol/L),甲状腺素(T4)为13.41 pmol/L(13.9~22.1 pmol/L);PTH为 74.90 pmol/L(1.6~6.9 pmol/L);胰岛素样生长因子-1(insulin-like growth factor 1,IGF-1)为9.75 ng/mL(60~350 ng/mL)。左手正位X片示:左手诸掌指骨粗短,骺线显示不清,部分骨骺弯曲、变形,见图3。双足正位X片示双足诸骨密度减低,骺线略模糊,部分骨形态不规则,见图4。双髋及双膝关节正位X片示双股骨远端、胫骨近段骨骺及干骺端形态欠规则,密度欠均匀,骺线模糊;双膝关节密度减低;双侧股骨干增粗并较短,股骨头增大,股骨颈增粗,沈通氏线欠连续;骨盆诸骨密度减低,见图5。脊柱X片示胸腰段序列如常,生理弯曲变直,椎体骨质密度略减低,各椎间隙尚均匀,见图6。颅脑磁共振示颅骨板障增厚,见图7。
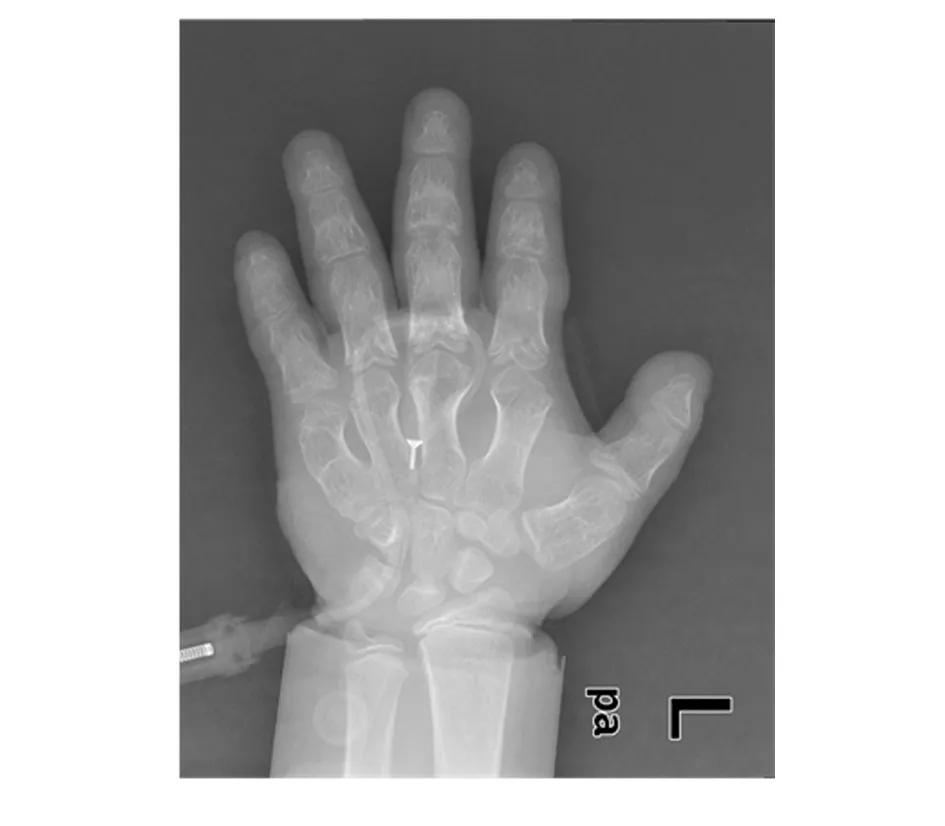
图3 患儿左手正位X片
治疗:入院后给予静脉补充葡萄糖酸钙,口服迪巧治疗,1 d后复查血钙1.57 mmol/L、血磷2.12 mmol/L;继续静脉补充钙剂,维生素D治疗,入院第3天再抽搐1次,表现同前,后未再抽搐。住院6 d后患儿出院,出院前复查血钙1.51 mmol/L、血磷2.83 mmol/L,嘱出院后继续口服迪巧0.3 g tid,钙锌口服液1支 bid,骨化三醇软胶囊0.25 μg qd及优甲乐12.5 μg qd治疗,定期复查。
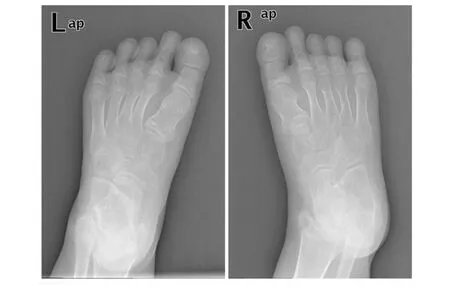
a.左足正位;b.右足正位。
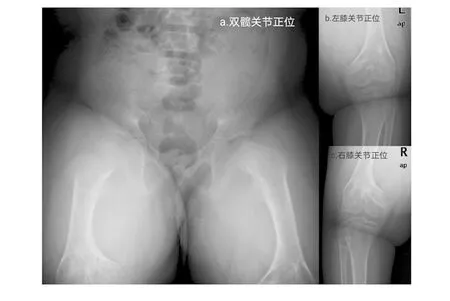
图5 患儿双髋及双膝关节正位X片
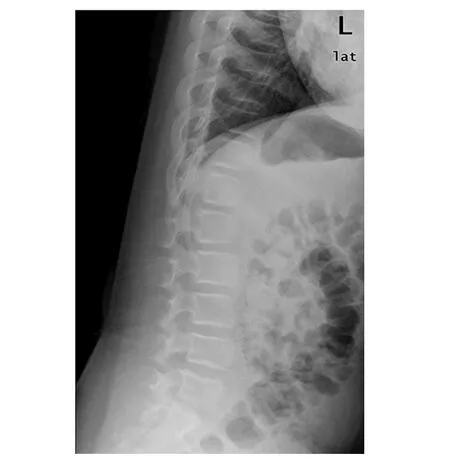
图6 患儿脊柱X片
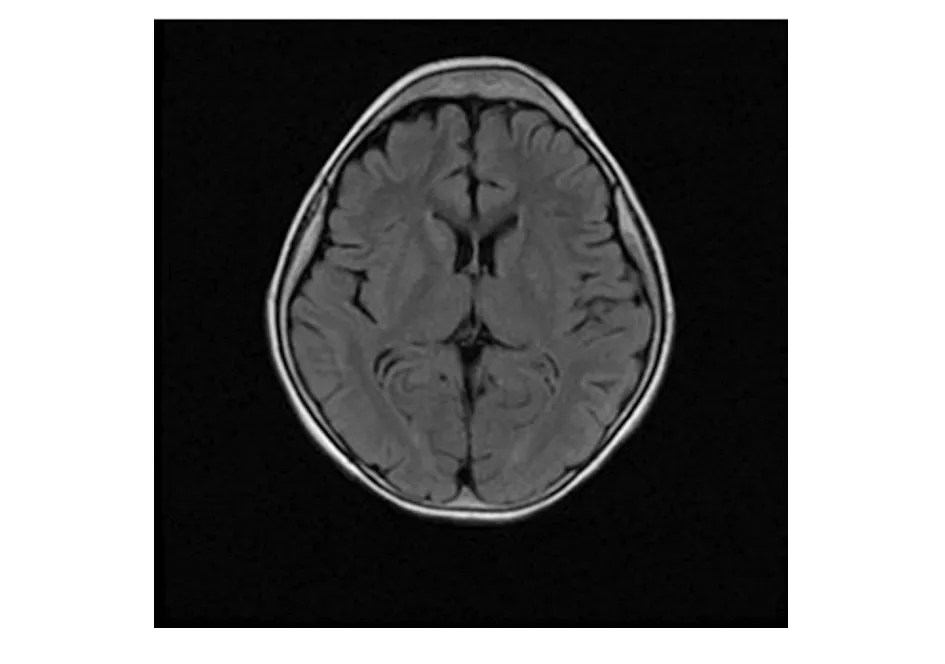
图7 患儿颅脑磁共振图像
随访:随访10个月,2019年8月26日复查TSH为16.090 mIU/L,T3为5.32 pmol/L,T4为11.83 pmol/L,PTH为53.42 pmol/L,血钙为2.13 mmol/L,血磷为2.29 mmol/L,25-羟维生素D为27.74 ng/mL;2019年10月13日复查TSH为11.580 mIU/L,T3为5.82 pmol/L,T414.01 pmol/L,PTH为68.23 pmol/L,血钙为2.18 mmol/L,血磷为2.30 mmol/L,后患儿未再复查血,未再出现抽搐。
2 基因检测
住院期间患儿家属同意行假性甲状旁腺机能减退综合征GNAS基因测序,采集患儿外周血,外送金域临床检测中心进行基因检测,采用一代测序方法,对GNAS基因的编码区进行直接测序,与参考序列NG_016194.1、NM_001077488.2进行比较,从而发现可能存在的基因突变。所检测到的基因变异见表1,所检测到的基因变异的解释:该突变为框内缺失突变(预计会使所编码蛋白质从第157位Glu发生缺失,但不会引起移码)。

表1 患儿GNAS基因测序检验结果
3 讨论
PTH抵抗是PHP的主要致病原理,根据注射PTH后尿液中cAMP是否升高,分为Ⅰ型(不升高)和Ⅱ型(升高)[1]。其中Ⅰ型又分为3型,Ⅰa型与编码GSα的GNAS 基因外显子突变有关,目前发现1-13号外显子都有可能发生突变[2-3],GSα活性降低,主要表现为AHO畸形、机体发育障碍及PTH抵抗,常伴有智力发育迟缓等,部分患儿可合并多种激素抵抗,最常见的是甲状腺功能减退。Ⅰb 型与GNAS启动子的甲基化有关,一般无AHO畸形及多种激素抵抗,主要特征是肾脏对PTH的抵抗。Ⅰc型分子机制不详[4-6],常有与Ⅰa型类似的AHO畸形和对多种激素抵抗的表现,但GSα活性正常,可能为Ⅰa型的变异[1,7]。但是在Mantovani等人[8]的研究中,对40例具有AHO畸形、从临床表现上诊断为PHPⅠa型的患者行基因检测发现,其中24例均无GNAS1基因1-13号外显子的突变,而是具有PHPⅠb型患者特有的GNAS1基因DMR区域的甲基化异常。
根据患者特殊的AHO体貌,结合低钙血症、高磷血症和过高的PTH水平即可诊断PHP[1],GNAS基因检测是诊断PHP的金标准[9]。
GNAS基因外显子发生突变是PHPⅠa型的主要发病机制,GNAS基因定位于20q13.3,包含13个外显子,是一个复杂的印迹表达基因,编码多种基因产物,GSα是其编码的最主要产物,该基因的变异类型多不一致,包括错义突变、无意突变、缺失突变,和新发现的插入突变、复杂突变、剪切位点突变[2-3],其中位于第7外显子的4bp的缺失(c.565-568delGAcT)是一个热点突变,在所报道的突变中大概约占17.4%[10]。正常情况下,甲状旁腺分泌的PTH与靶器官细胞上的受体结合,激活与受体耦联的鸟苷酸结合蛋白,即G蛋白,进一步激活腺苷酸环化酶(AC),AC在镁离子的帮助下,将ATP转化为cAMP,cAMP作为第二信使进一步激活蛋白激酶A(PKA),导致蛋白磷酸化,产生生物学效应[11]。目前的研究显示,PHP患者中,GNAS基因突变导致了PTH的受体蛋白(GSα蛋白)活性下降,使cAMP-蛋白激酶通路受阻,当PTH刺激靶细胞时,不能产生相应的生理效应,并且由于代偿,甲状旁腺增生肥大,PTH合成与分泌增加[12-13]。该患儿染色体突变位置为20q13.3,经基因检测其GNAS基因6号外显子有一缺失突变,与其他突变不同,该突变导致1个氨基酸的完整缺失,并未引起移码突变, Long DN等人[14]的研究中曾提及该突变:ex.6: c.469_471(E157del),与该患儿所鉴定的突变为同一密码子,引起了同一氨基酸的缺失,即编码Gsα的第157位谷氨酸的缺失,从而导致了疾病的发生。
综上所述,本例报道鉴定了1个导致PHP的基因突变,即ex.6: c.470_472(E157del)。
