二萜生物碱化学研究的回顾
2021-09-01王锋鹏
王锋鹏
四川大学华西药学院天然药物系,成都 610041
二萜生物碱是一类重要的天然化合物。由于生物多样性、结构的复杂性以及有趣的化学和重要的生物活性,二萜生物碱的研究长久来一直受到人们的重视。特别是近30年来,由于色谱与波谱技术的发展与应用,发现的二萜生物碱的数目急剧增加。截止2019年底为止,发现的二萜生物碱已逾1 500个。而绝大多数二萜生物碱分自于毛茛科乌头属和翠雀属,以及蔷薇科绣线菊属。仅仅极少数散在分布在其他植物中。我们先后比较系统地归纳总结C20-、C18-和C19-二萜生物碱的诸多方面,主要包括结构分类、分布、生源关系、存在、谱学和单晶X-射线衍射分析、化学性质与转化合成,生物活性,以及对1998~2008年期间报告的二萜生物碱的植物化学、合成和生物活性的综述[1-5]。本文仅简要地回顾我们研究组过去30多年来,在二萜生物碱化学方面的主要研究结果,供同行们指正。
1 化学成分和结构分类的研究
对36种乌头属和翠雀属植物做了比较系统的化学研究。从中分出新化合物约170个,新骨架6个。其中,racemulosine(1)是一种罕见而奇特的C20-二萜碱。其特点是分子中A/B/C三个环生源上由前体物denudatine-type二萜碱,经A-nor/B-homo/C-nor重排而来。同时,C-17上引入氨基[6]。首次分得海替烷型(hetidanes)二萜化合物campylopin(2),生源上可认为是海替定二萜生物碱的前体化合物[7]。新型阿苦塔型(arcutanes)二萜化合物atropurpuran(3),其结构呈罕见的五环笼状结构,生源上可认为是阿苦塔型二萜生物碱(arcutines)的前体物[8](见图1)。
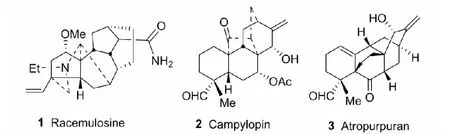
图1 发现的新骨架化合物Fig.1 Compounds with new skeletons identified during our studies
紫乌定(episcopalidine,4)[9,10]是从云南产苍山乌头(Aconitumcontrodum),最初误定为紫乌头(A.episcopale)中分出。在当时条件下,其结构测定颇费周折。困难的是,分子中两个酮基(C-6、C-13)的弛豫时间T1不同,导致通常情况测不出C-6信号。同时,影响酯基偕碳质子δ值的两种矛盾的因素(空间效应:H-2(e) >H-3(a);诱导效应:H-C-OBz >H-C-OAc)(见图2A),使其定位难以判断。此外,跨环效应的存在,也使非充分碱化条件下,测得的C-6信号的δ值偏离真值。最后,除NMR和辅助化学法外,结构的确证是由单晶X-射线分析完成的[9,11]。
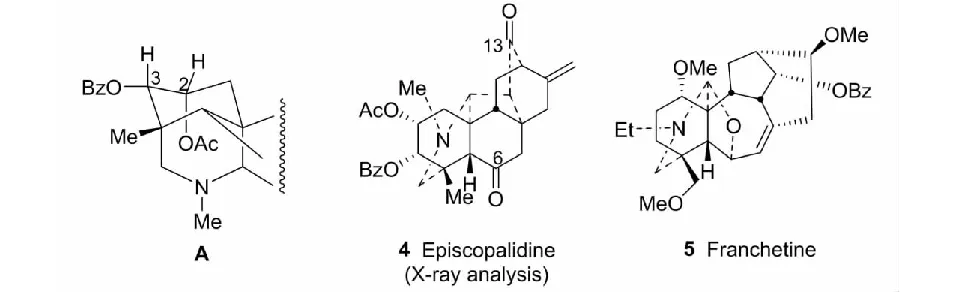
图2 Episcopalidine的结构研究及franchetine的结构式Fig.2 Study on structure of episcopalidine and the chemical structure of franchetine
修正了46个二萜生物碱的结构[12-17]。其中franchetine(5)又是一种新骨架的化合物。修正涉及酸-碱反应,结构与构象,含特殊结构单元(如季胺碱,氮杂缩醛、具跨环效应)生物碱的成盐,跨环效应,二萜生物碱的光谱特征与特有的化学反应,影响F3CCOO-中C-13信号强度的因素,以及光谱技术(19F NMR、1H-15N NMR、CSI-MS)和单晶X-射线分析等[15,16]。
在二萜生物碱的结构分类中,S.W.Pelletier 做了许多奠基性的工作[2,4]。1983年我们最早建议从C19-二萜碱中将C18-二萜碱分出[9]。2002年我们首次提出以骨架类型结合氮原子为中心的4级(类型、型、亚型、组)的分类方法,将复杂的C20-二萜碱分为4大类(乌头烷类、考烷类、重排类,双二萜碱类)20型35亚型43组[2]。自后,又依次对C18-和C19-二萜生物碱的结构分类进行归纳总结,分别将C18-和C19-二萜生物碱分为2类型4亚型和6类型13亚型19组[3,4]。
2 化学反应的研究
二萜生物碱是一类具有多环笼状复杂结构的化合物。无论是结构修饰,还是合成研究,都常常涉及多种功能基的转换与键的裂解与形成,以及骨架的转化等。二萜生物碱的骨架源于四环二萜,则更使其化学反应复杂多样。这里,汇集的化学反应都是我们研究此类生物碱,尤其是转化合成紫杉烷类研究中所遇到的反应。兹择主要者,加以概述。
2.1 N-去乙基化与亚胺形成
这是A环修饰的关键反应之一。文献报告的方法较多,但产物收率低或反应后处理复杂。经过多次摸索,我们发现一种新的去氮乙基方法,来制备去氮乙基物和亚胺化合物(见图3)[18]。其特点是反应条件温和、收率高,且可通过改变反应条件人为地控制产物,此法更适用于乌头碱型二萜生物碱[19]。该法成为后来候选抗心衰药物中乌宁工业生产的重要反应之一。
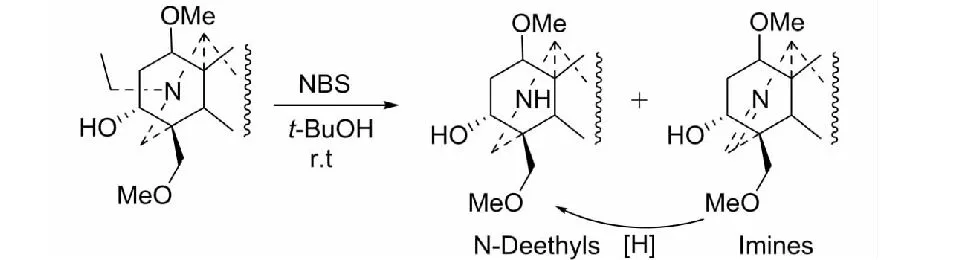
图3 NBS法制备去氮乙基物和亚胺化合物Fig.3 Preparation of deethylated amines and imines using NBS
2.2 键的形成与裂解
2.2.1 N-C(6)键的形成
文献报道真空热解牛扁酰胺酸(6),仅得脱羧物。我们的研究发现在类似条件(15 mmHg,220 oC,25 min)下,由底物6,经一系列反应(Pinacol重排、SN2取代、脱羧、Grob裂解)生成含N-C(6)键的化合物6a。同时,伴随N-C(19)键的裂解。这是一个异常的裂解C(19)-C(4)键,而形成N-C(6)键的反应。化合物6a再经氢解,则生成裂解N-C(6)键的化合物6b和6c[20](见图4)。
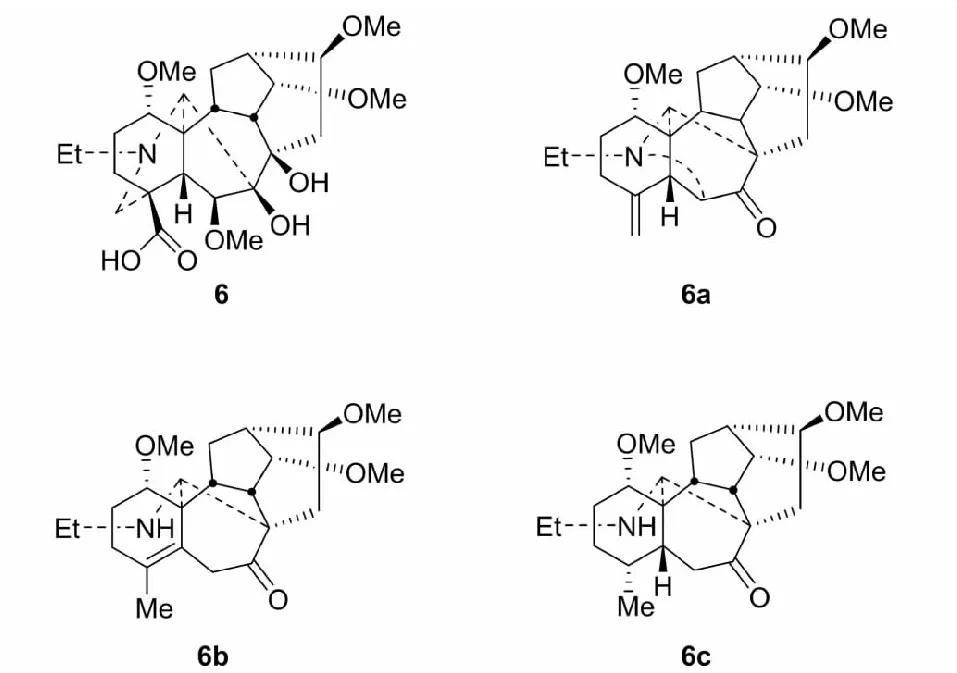
图4 牛扁酰胺酸6及其裂解产物6a/6b/6cFig.4 Structures of 6 and its cleavage products 6a/6b/6c
2.2.2 C(7)-C(17)键的裂解
文献有多种方法报告,但操作繁冗、收率低[4]。我们先后发现N-氧化物热解法(见图5A)[4,21]、应用N-氯物重排(见图5B)[4]和8-氯物热解还原法(见图5C)[4,22,23],尤其后者反应收率高达80%~90%,是一种新的更实用的方法。而在diglyme中热解N-氧化物反应,则经历了一个邻基参与的复杂的过程[21]。
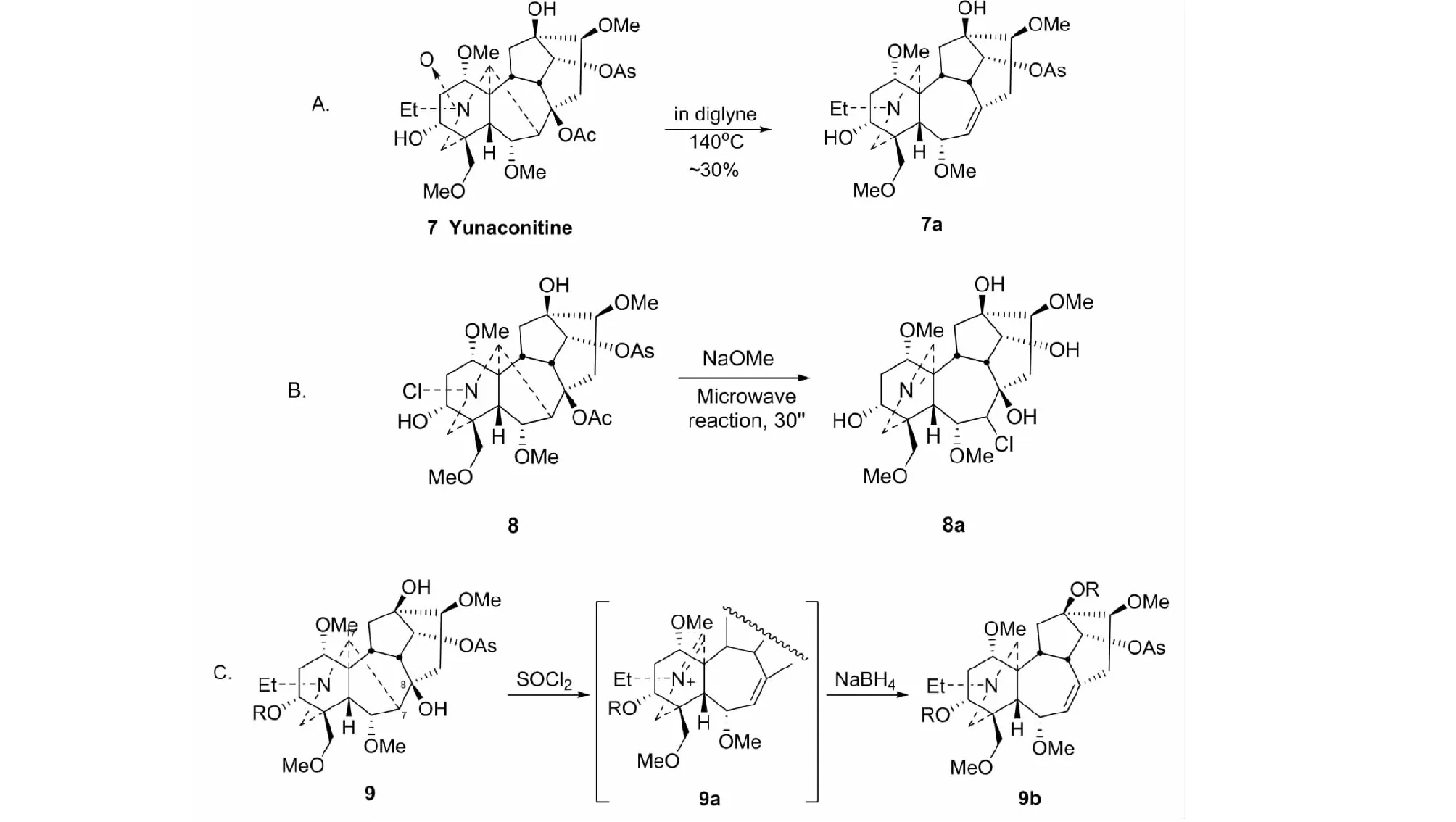
图5 三种C(7)-C(17)键裂解的方法Fig.5 Three methods for breaking C(7)-C(17) bonds
在转化合成内酯型二萜生物碱heteratisine中,我们又偶然发现,在真空热解条件下,由N-去乙基-8-乙酰氧基-C19-二萜生物碱,如化合物10和化合物11可分别中等收率地制得7,17-次裂产物10a/10b和11a[24](见图6)。这是一种新的裂解C(7)-C(17)键的方法。但不适用于含13-OH基的底物。
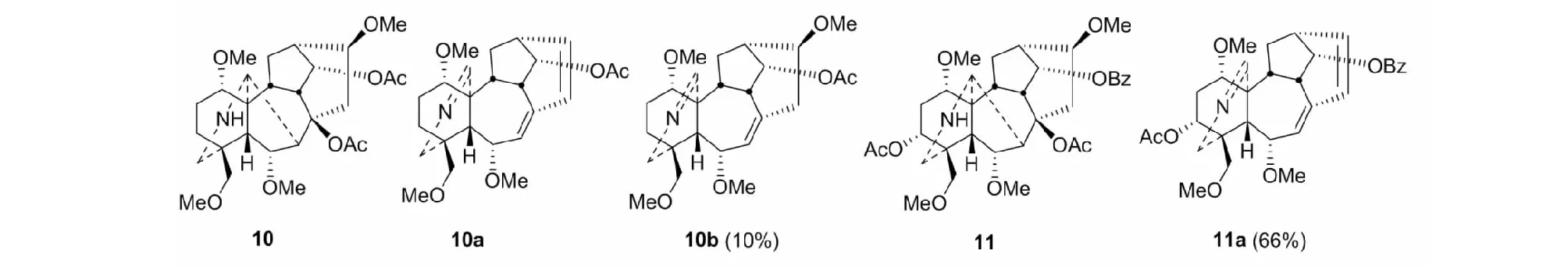
图6 7,17-次裂产物10a/10b和11aFig.6 7,17-Seco products 10a/10b and 11a
2.2.3 C(9)-C(10)键的裂解
在我们试图由化合物12于NaOH-DMF液中,经Wagner-Meerwein重排裂解C(9)-C(10)键时,却得到9,10-次裂产物12a。缩短反应回流时间至4h时,才得到12b(见图7)。显然,二者都是Grob裂解的产物[25]。这个反应成为后期转化合成紫杉烷类似物的关键步骤之一。
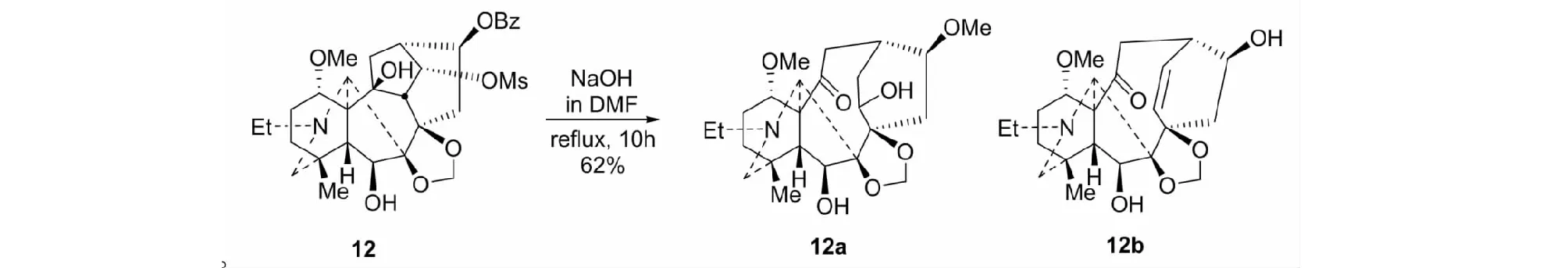
图7 9,10-次裂产物12a/12bFig.7 9,10-Seco products 12a/12b
2.2.4 C(12)-C(13)键和C(12)-C(14)键的裂解
文献未见报告。经过种种努力,我们终于发现在十分温和条件下,C19-二萜生物碱B经semipinacol重排,可高收率地得到的重排物C,C再与Br2或SOCl2反应即得所希望的12,13-次裂C19-二萜生物碱D(见图8)[26]。
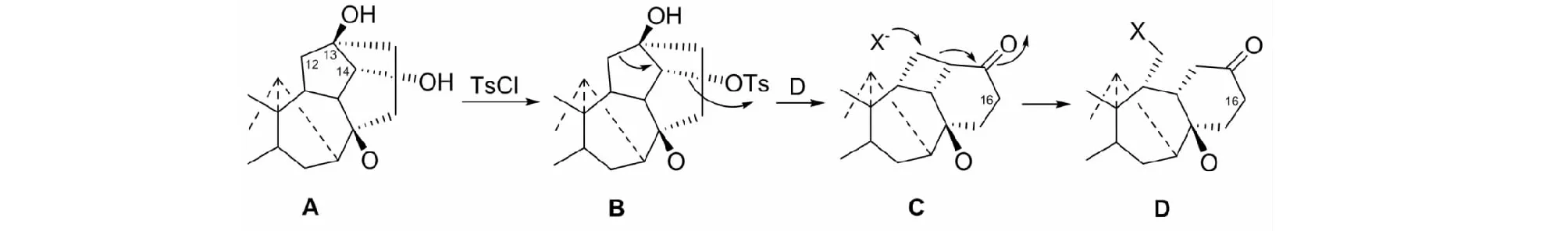
图8 化合物B的重排产物C和12,13-次裂产物DFig.8 Rearrangement from compound B to C and 12,13-seco product D
以滇乌碱(yunaconitine,7)为原料,经Jone’s氧化/碱化,C(14)位羟基选择性甲基磺酰化,再与DMF-NaOH(150 ℃,10 h)反应即生成一对差向异构的12,13-次裂化合物13和14(总收率85%)[27]。类似地,应用A环不同的底物时,也高收率地获得12,13-次裂产物(见图9)[26]。
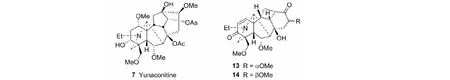
图9 12,13-次裂产物13和14Fig.9 12,13-Seco products 13 and 14
2.3 扩B环
当我们试图用文献方法[4],由亚胺15与NaNO2-HOAc反应脱氮时,却意外地得到扩B环的二萜化合物15a和15b[28]。更重要的是,底物有扩展性(见图10)[29,30]。
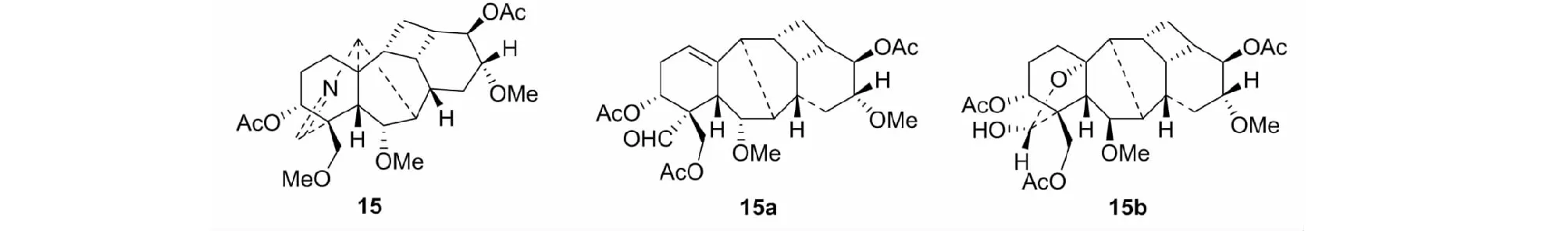
图10 扩B环的二萜化合物15a/15bFig.10 Diterpenoid derivatives 15a/15b with expanded B ring
类似地,亚胺16或17与NaNO2-10% H2SO4-THF反应,得扩B环的同一二萜物18(Scheme5)[31]。该法成为另一条新的转化合成紫杉醇类似物的关键反应(见图11)。
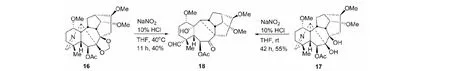
图11 扩B环的二萜化合物18Fig.11 Diterpenoid compound 18 with expanded B ring
2.4 跨环效应
跨环效应仅在某些特定结构的天然产物中存在。日本学者Kakimoto和Pelletier[4]最早指出海替定型C20-二萜生物碱可能存在这种效应。我们应用碳谱首次证明海替定型紫乌定类似物存在跨环效应(见图12)[11,32]。后来,又证实vakognavine-type C20-二萜生物碱分子中,如trifolialasines D(19)、E(20)、F(21)等也存在跨环效应[16,33]。
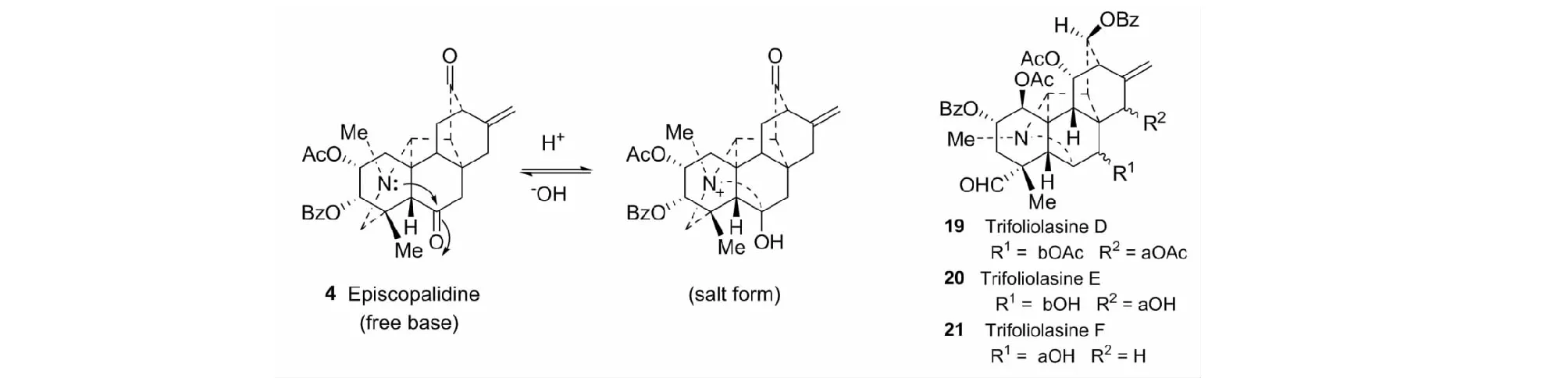
图12 二萜生物碱中的跨环效应Fig.12 Transannular effect in diterpenoid alkaloids
2.5 重排反应
雪上一支蒿甲素(光翠雀碱,denudatine,22)是药用草乌类药材质量控制的重要特征性成分,我们对其理化性质进行了比较详细地研究。结果发现了光翠雀碱与10% HCl甲醇液反应,生成有趣的3个重排反应产物及其重排产物之间的互相转化(见图13)[34]。
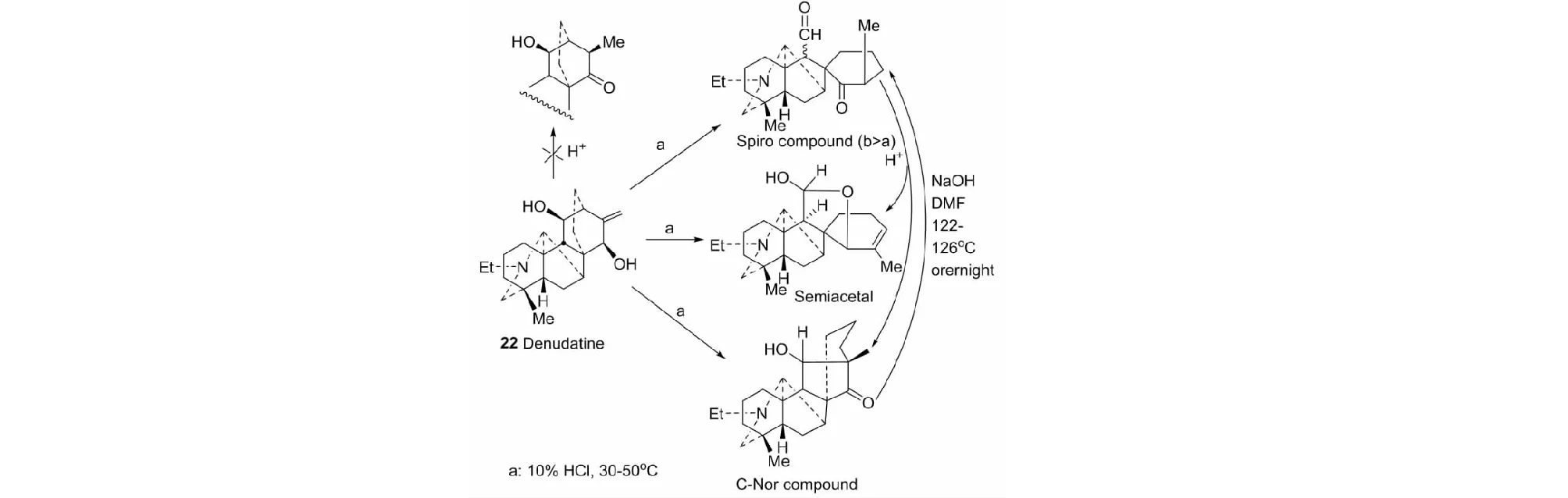
图13 光翠雀碱的重排产物及其相互转化Fig.13 Rearrangement products of denudatine and their interconversion
当我们试图使亚胺23季胺化来裂解N-C(19)键时,在5% NaOH甲醇室温条件下,却意外地得到一个A环重排的新骨架C19-二萜碱23a[35]。这是一个新奇的重排反应,反应过程比较复杂。有趣的是,化合物24在不同的反应条件下,均生成同一化合物24a(见图14)[36]。重排反应涉及N-C(19)键的裂解,以及N-C(6)键和δ-内酯的形成。
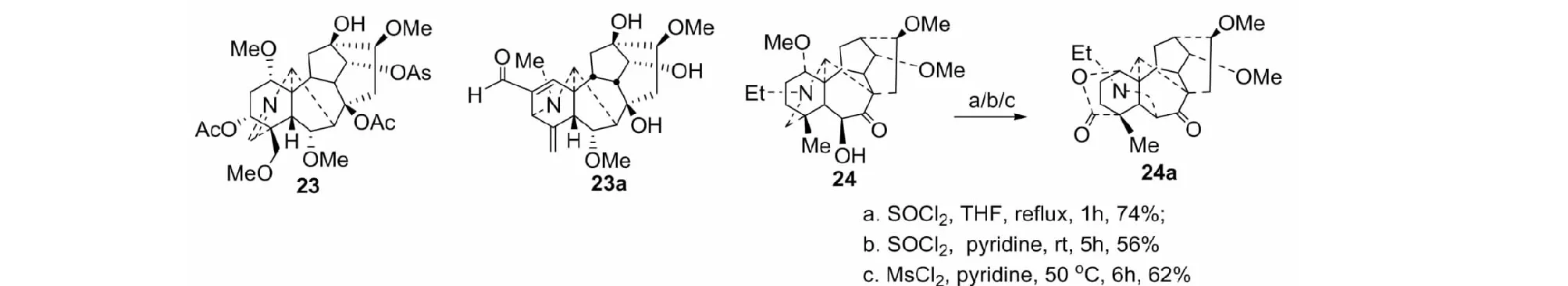
图14 化合物23与24的重排Fig.14 Rearrangement reactions of 23 and 24
2.6 复杂二萜的形成
含八元环B的重排aconane二萜25与Br2反应时,并未得到期望的8,17-seco产物,而生成25a和异常复杂的溴化物25b(见图15)。25b的生成可能涉及包括连续的3次W.M.重排以及溴化、消除和氧化的反应过程。但当25与(Boc)2O反应时,却生成化合物25c、25d和25(见图15)。化合物25c和25d的生成也可能经连续的W.M.重排,以及酰化过程[31]。
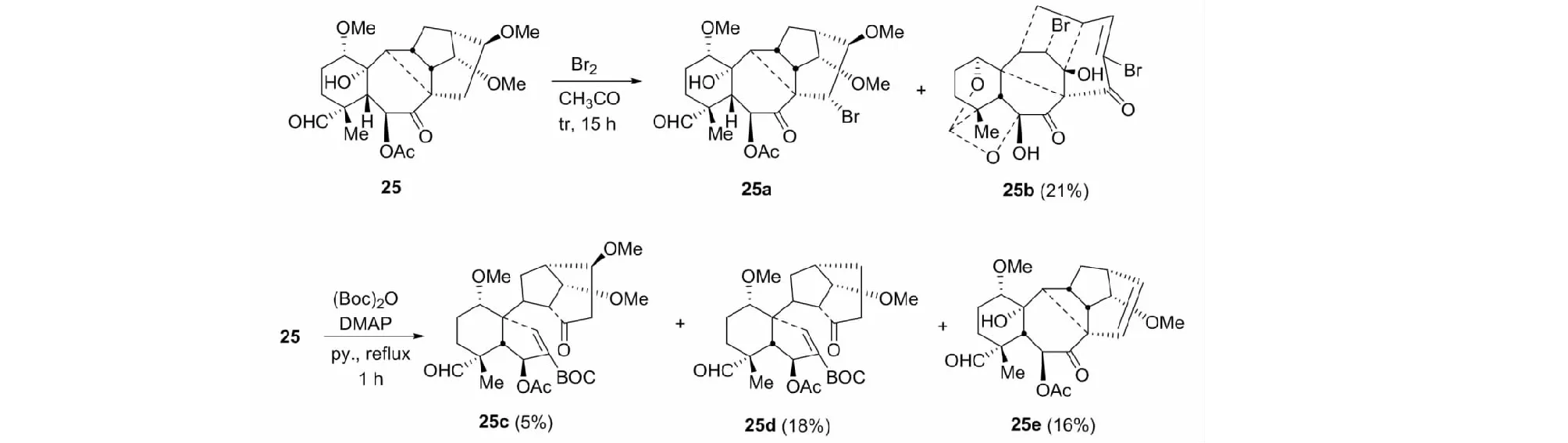
图15 复杂二萜化合物25a~e的形成Fig.15 Formation of complex diterpenoid compounds 25a-e
2.7 海替定型与阿替生型C20-二萜生物碱的互相转化
在梁晓天教授指导下,经种种探索,终于利用海替定26分子中,2α-直立羟基捕获中间体的方法来裂解C(20)-C(14)键,而转化成阿替生型生物碱(见图16)[37]。
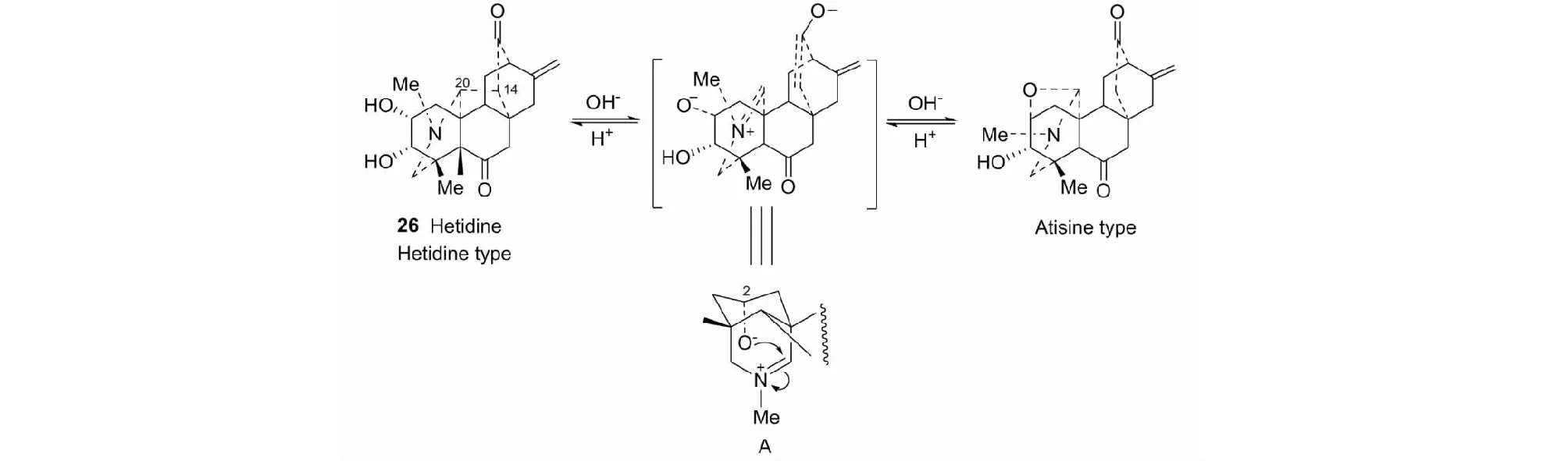
图16 海替定型与阿替生型C20-二萜生物碱的互相转化Fig.16 The interconversion of hetidine- and atisine-type C20- diterpenoid alkaloids
3 转化合成紫杉醇类似物和紫杉醇类似物的合成
3.1 转化合成紫杉醇类似物
在抗癌药紫杉醇(taxol®,27)和多烯紫杉醇(docetaxel®,28)的研究开发成功背景下,经过思考与联想,于90年代初我们提出由丰富存在的二萜生物碱,如滇乌碱(yunaconitine,7)、印乌碱(indaconitine,29)和deltaline(30)等为原料,转化合成紫杉醇的战略。10多年来,先后提出6个战略进行研究[4]。
最后,如图17所示,以deltaline(30)为原料,经6步反应(O-去甲基化/内酰胺化、碱水解,BzCl选择性保护/甲磺酰化,Grob裂解/O-甲基化保护,O3裂解/O-甲基化保护,B2H6还原和Pelletier’s裂解),以总收率16%合成紫杉烷类似物35。化合物35经NaBH4还原、乙酰化和m-CPBA/H+氧化制得类似物36(收率69%)(见图17)[38]。
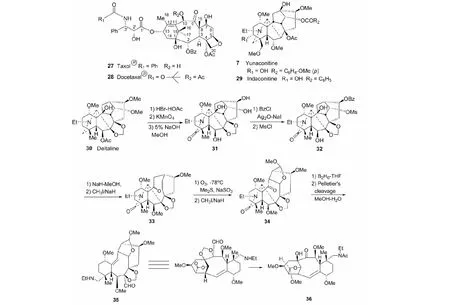
图17 Deltaline转化合成紫杉醇类似物35和36Fig.17 Conversion synthesis of taxol analogues 35 and 36 from deltaline
另外,利用扩B环的关键反应,以牛扁碱(lycoctonine,37)为原料,如图18所示路线,最终也完成转化合成紫杉醇类似物A/B/C母核的研究[30]。该法优点是步骤少,不足的是最后一步收率低。
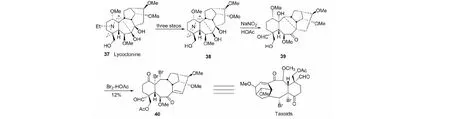
图18 牛扁碱转化合成紫杉醇类似物Fig.18 Conversion synthesis of taxol analogues from lycoctonine
3.2 紫杉醇类似物的合成
在完成紫杉醇类似物母核后,我们以deltaline(30)为原料,合成了多烯紫杉醇类似物41和42(见图19)[39]。转化中难点是13α-酯基的引入。遗憾的是,这两个化合物均无细胞毒活性。
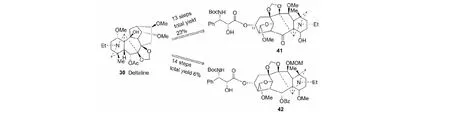
图19 多烯紫杉醇类似物41和42的合成Fig.19 Synthesis of docetaxel analogue 41 and 42
4 多烯紫杉醇D环修饰及其构效关系的研究
由于作为转化合成的活性目标物中,四元环氧结构既必需,又不稳定与难于合成。而文献对D环修饰未有突破。所以,我们决定对紫杉醇类分子中D环进行修饰。通过仔细分析,我们设计以10-deacetyl baccatine III(43)为原料,用10步反应总收率1.3%完成含γ-内酯化合物44的合成(见图20)。44对多种肿瘤细胞抑制作用与多烯紫杉醇相当,其对人黑色素瘤的抑制作用是多烯紫杉醇的2倍[40]。这是紫杉醇类D环修饰的一个新突破,从而大大简化了转化合成的步骤与难度。
在制备化合物44时,需选择性由酰基迁移开裂原料10-deaxeryl-baccatin III(43)分子中D环。我们发现10-deacetyl-baccatine III与TiCl4反应,可高收率(66%)地制得关键中间体45(见图20)[41]。这是一个新的选择性开裂紫杉醇类D环的方法。
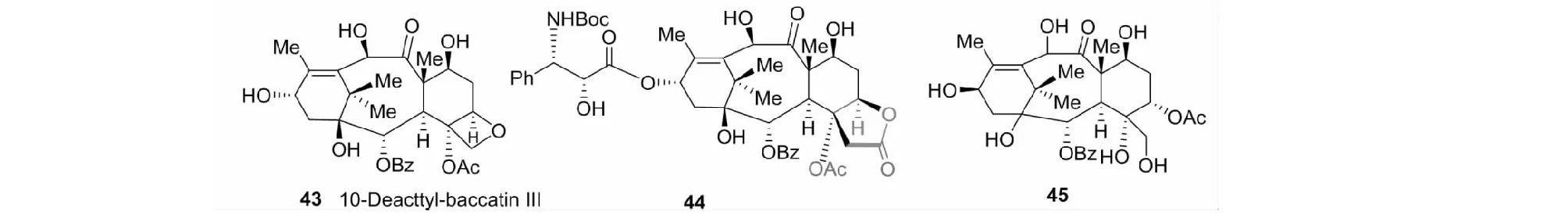
图20 多烯紫杉醇D环修饰化合物44及45Fig.20 Docetaxel D-ring modified compounds 44 and 45
5 催化氢化还原醛与六元环酮的新方法
醛和酮的还原是有机化学和天然产(药)物化学的常见反应。还原剂多为金属氢化物如NaBH4、LiAlH4等。文献未报告用氧化铂-三乙胺常温常压催化氢化来还原醛或酮的。我们在紫杉醇构效关系研究中,偶然发现在PtO2(5 mol%)-Et3N(1当量)-95%乙醇(溶剂)条件下,常温常压催化氢化种种醛(脂肪醛/芳香醛、链状或环状醛)以及包括复杂体系中的六元环酮还原成醇(见图21)[42]。显然,这是一种具有广泛应用价值的创新性的还原方法。

图21 氧化铂-三乙胺常温常压催化氢化还原醛或酮Fig.21 Hydrogenation of aldehydes or ketones catalyzed by PtO2-Et3N at normal temperature and pressure
6 二萜生物碱的合成
二萜生物碱结构复杂、活性多样,是最有趣最复杂的生物碱之一。其全合成的黄金期是上世纪60-90年代。近年来又成为全世界天然产物化学家全合成关注与研究的热点与难点领域之一。我们从2008年开始,进行二萜生物碱全合成和复杂多样性二萜生物碱合成的研究,并取得如下突破性的进展。
6.1 环系的构建
创新性地完成A/E、C/D、A/E/F、B/C/D和A/B/E/F环系的构建。
6.1.1 A/E环系的构建
在二萜生物碱中,含5-OH羟基者十分罕见,如nordhagenil A(46)。以cyclohex-2-enone (47)为原料,经9步总收率7%合成具A/E环系的化合物51(见图22)[43]。
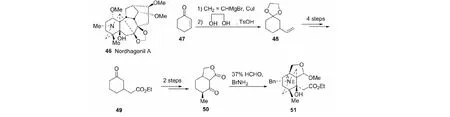
图22 具A/E环系化合物51的合成Fig.22 Synthesis of compound 51 with A/E rings
6.1.2 C/D环系的构建
以芳醛52为起始原料,经关键中间体53,共5步反应合成高度官能化的化合物54。其C/D环系与C20-二萜生物碱中相对应(见图23)[44]。
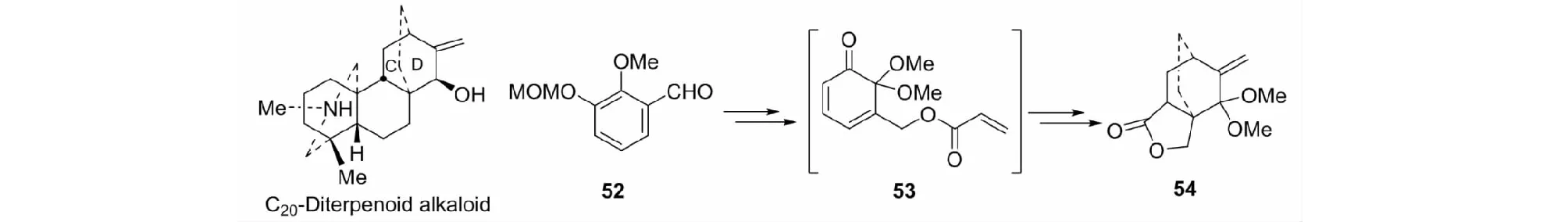
图23 具C/D环系化合物54的合成Fig.23 Synthesis of compound 54 with C/D rings
6.1.3 A/E/F环系的构建
以目标化合物royleinine(55)为准发展了两种方法,其一是以56和57为原料进行DA反应生成关键中间体61和62,共11步反应合成期望的化合物63(见图24A)[45]。59→60是经I+3介导的分子内氮环丙烷化完成的。另一种是以2,2-bis (3-methoxy-3-oxopropyl) malonic acid(64)为原料,经关键中间体67和68,共11步总收率15%合成目标物69(见图24)[46]。
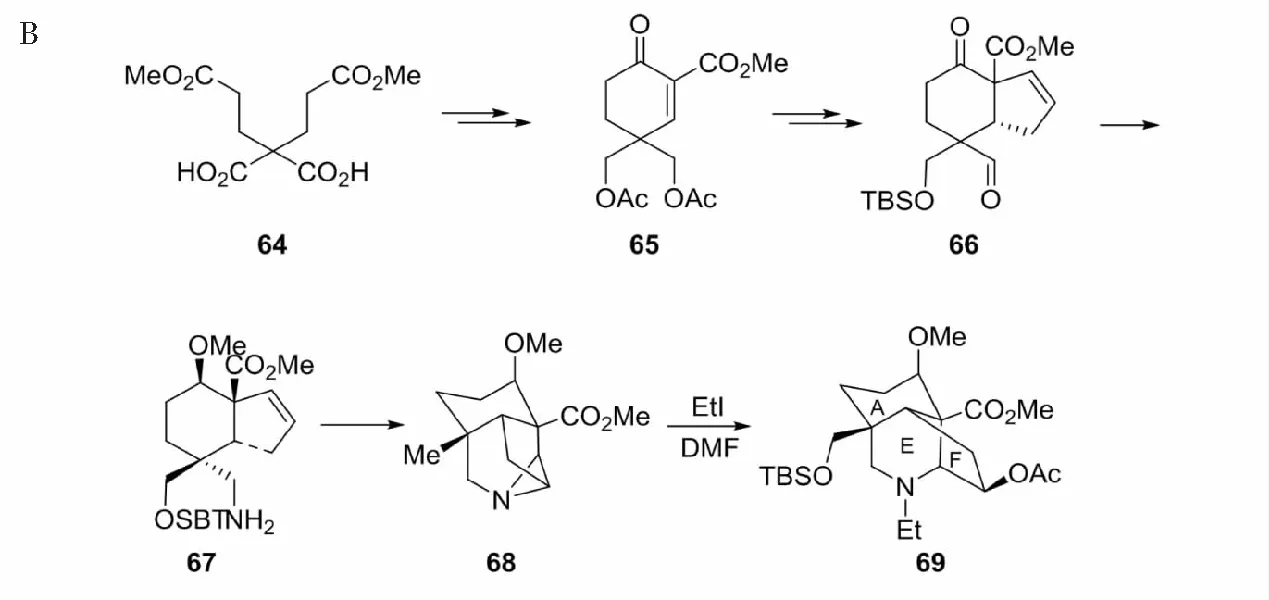
图24 具A/E/F环系化合物63和69的合成Fig.24 Synthesis of compounds 63and 69 with A/E/F rings
6.1.4 Racemulosine的合成:A/E/F环系的构建
如前所述,racemulosine(1)是我们报告的一个新骨架的C20-二萜生物碱。但除A环为五元环以及骨架碳为C20外,与C19-二萜生物碱很相似。我们以易得的二醇70为原料,经关键中间体71和72,共18步合成目标物76(见图25)[47]。
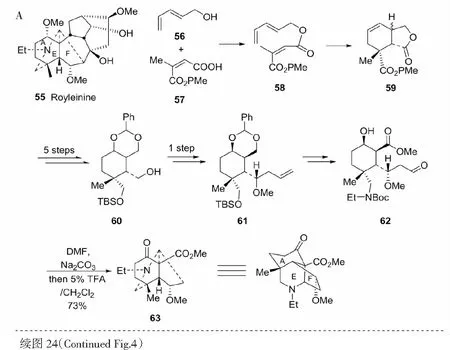
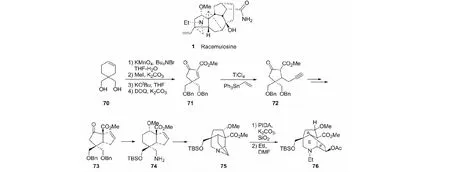
图25 Racemulosine的化学结构及其A/E/F环系的合成Fig.25 Chemical structure of racemulosine and construction of its A/E/F rings
6.1.5 B/C/D环系的构建
此环系构建是以乌头碱(aconitine,77)和isodelphine(78)为最终目标物而设计的。以2-methoxyphenol THP ether(79)为原料,经关键中间体83而合成目标分子84(见图26)[47]。
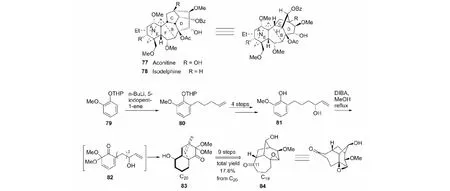
图26 具B/C/D环系化合物84的合成Fig.26 Synthesis of compound 84 with B/C/D rings
6.1.6 A/B/E/F环系的构建
以franchetine(85)和consolarine(86)为最终目标物,构建A/B/E/F模拟研究,以methylcyclohept-1-enecarboxyclate(87)为原料,经关键中间体90和91,合成期望的化合物94(见图27)[49]。
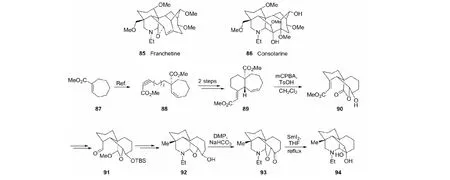
图27 具A/B/E/F环系化合物94的合成Fig.27 Synthesis of compound 94 with A/B/E/F rings
6.2 全合成
6.2.1 仿生合成Heteratisine
Heteratisine(95)具有显著的抗心律失常作用,但天然来源困难。通过反复研究,我们以deltaline(30)为原料,以15步总收率3.1%完成其合成(见图28)[50]。
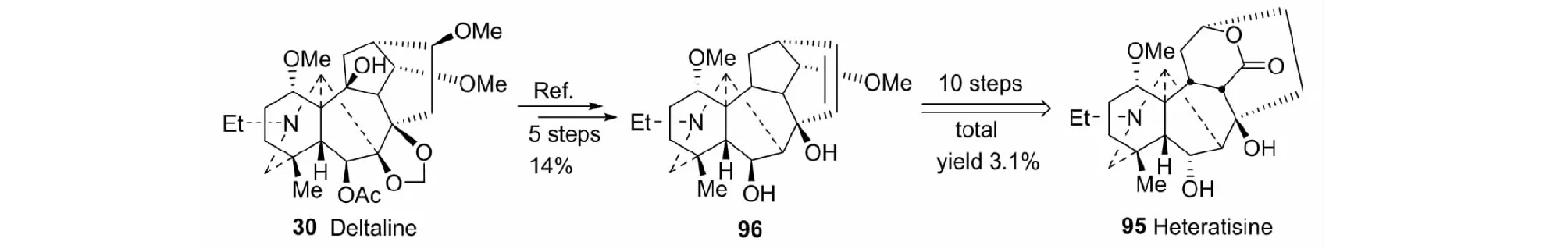
图28 Heteratisine的仿生合成Fig.28 Biomimetic synthesis of heteratisine
6.2.2 (±)-Atisine和(±)-Isoazitine的全合成
这是两个重要的C20-二萜碱。我们以邻甲酸乙酯环己酮97为原料,经18步完成这两种生物碱(±)-atisine(105)和(±)-isoazitine(106)的全合成(见图29)[51]。关键反应是全新的氧化去芳香化/分子内DA环加成。
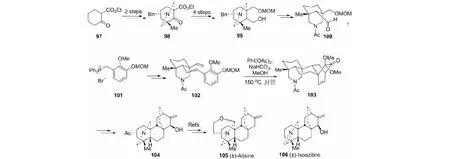
图29 (±)-atisine和(±)-isoazitine的全合成Fig.29 Total synthesis of (±)-atisine and (±)-isoazitine
6.2.3 (±)-Spiramilactone B、(±)-spiraminol、(±)-dihydroajaconine和(±)-spiramines C和D的全合成
这是两类化合物:阿替生烷型二萜和阿替生型二萜生物碱的首次合成(见图30)。关键反应(步骤)是:①串联逆Dids-Alder/分子内Diels-Alder顺序完成三环[6.2.2.0]环系构建(112→115);②高效非对映选择性的1,7-炔环异构化构建功能化的四环二萜阿替生烷(117→119);③由δ-内酯迁移成内酯(119→120);④在spiramilactone B(107)后面合成阶段,引入生源转化反应(108→109/110,121→109)[52]。
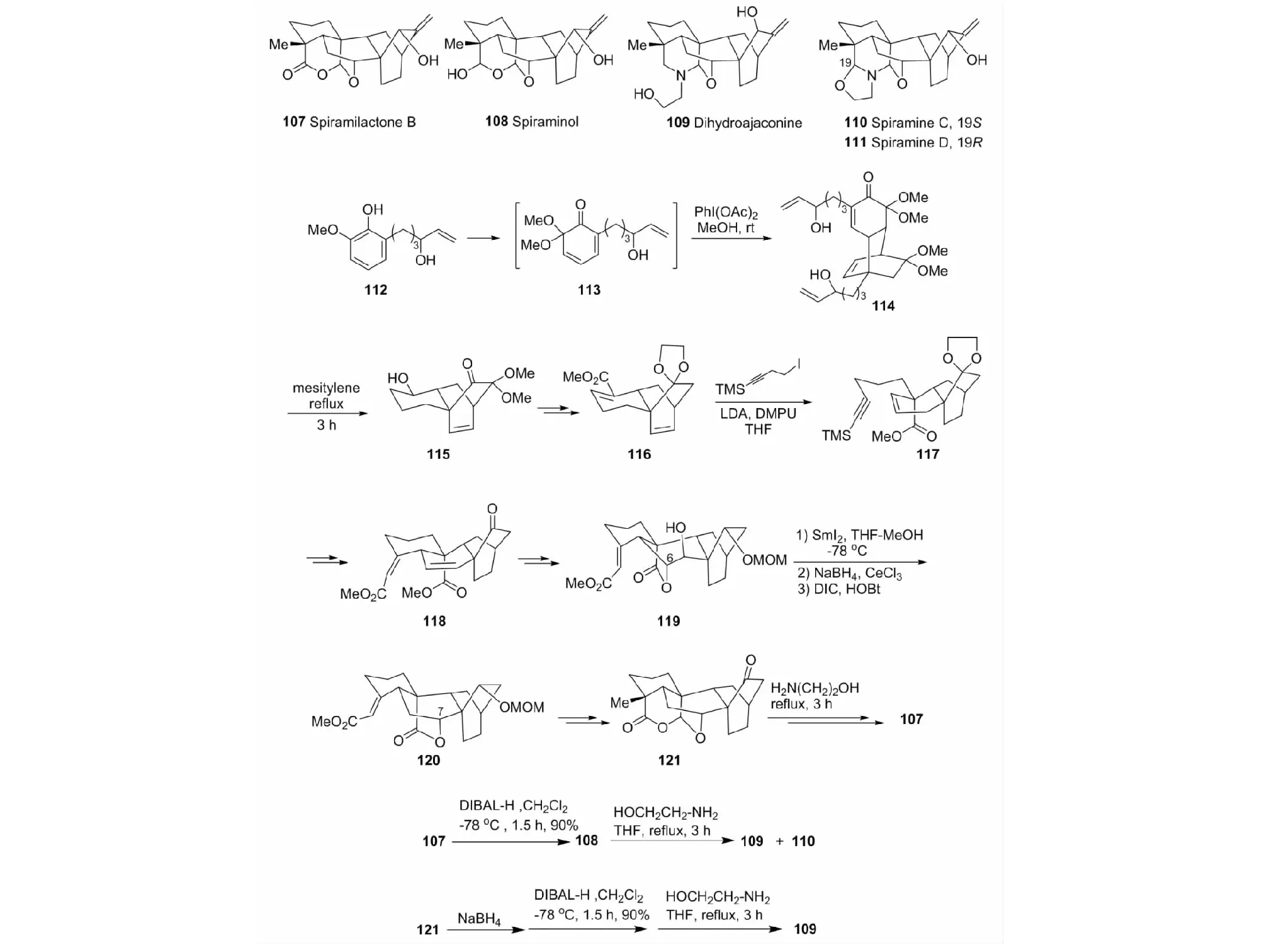
图30 (±)-Spiramilactone B、(±)-spiraminol、(±)-dihydroajaconine和(±)-spiramines C/D的全合成Fig.30 Total synthesis of (±)-spiramilactone B,(±)-spiraminol,(±)-dihydroajaconine and (±)-spiramines C/D
6.2.4 4种C20-二萜生物碱骨架和C20-二萜生物碱dihydroajaconine和gymnadine的全合成在前期研究的基础上,加之,我的年青同事—天然产物合成化学家秦勇教授的加入,我们以化合物122为原料,全合成了4种C20-二萜生物碱骨架(atisine-type、ajaconine-type、denudatine-type、heti-dine-type)和2种天然产C20-二萜生物碱gymnadine(132)和dihydroajaconine(133)(见图31)[53]。
如图31所示,再次应用Corey-Seebach反应和氧化去芳香化/分子内D-A反应由123和124合成了126(atisine-tytpe)。首次采用自由基酰胺氧化-分子内羟基捕获的方法,将128转变成化合物129(ajaconine-type)和130。又依Iwabuchi方法氧化131中羟基后,接着在SmI2介导下,发生aza-pinacol偶联生成化合物132。132引入15-羟基后就生成15-epi-gymnadine(133)和gymnadine(134,denudatine-type)。此外,129经一系列功能基的转换与引入后,完成了dihydroajaconine(135)全合成。最后,136经aza-prins反应生成hetidine-type的二萜生物碱化合物137。
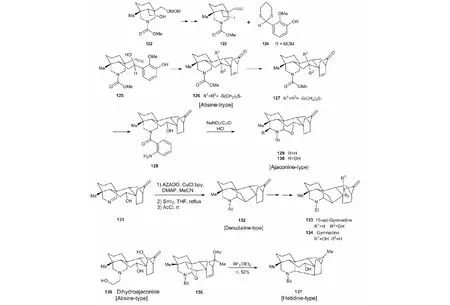
图31 4种C20-二萜生物碱骨架和C20-二萜生物碱dihydroajaconine和gymnadine的全合成Fig.31 Assembly of four types of C20-diterpenoid alkaloids and total synthesis of dihydroajaconine and gymnadine
6.2.5 复杂多样性二萜生物碱的合成
这是我们近几年开展的研究,目的是为新药的活性筛选提供基础。最先,以易得的deltaline(30)为原料,采用环变形途径(ring-distortion approach),以少于10步的反应,合成了约50个二萜生物碱的衍生物,其中代表性化合物如图32所示[54]。接着,又以talatisamine(148)为起始物,制备了20个具有新颖C/D环结构单元的二萜生物碱化合物。其代表性化合物如图33所示[55]。初步活性筛选表明,147对人乳腺癌MCF-7示出较好的活性(IC50=10.6 μM)。而化合物216具有突出的镇痛作用。
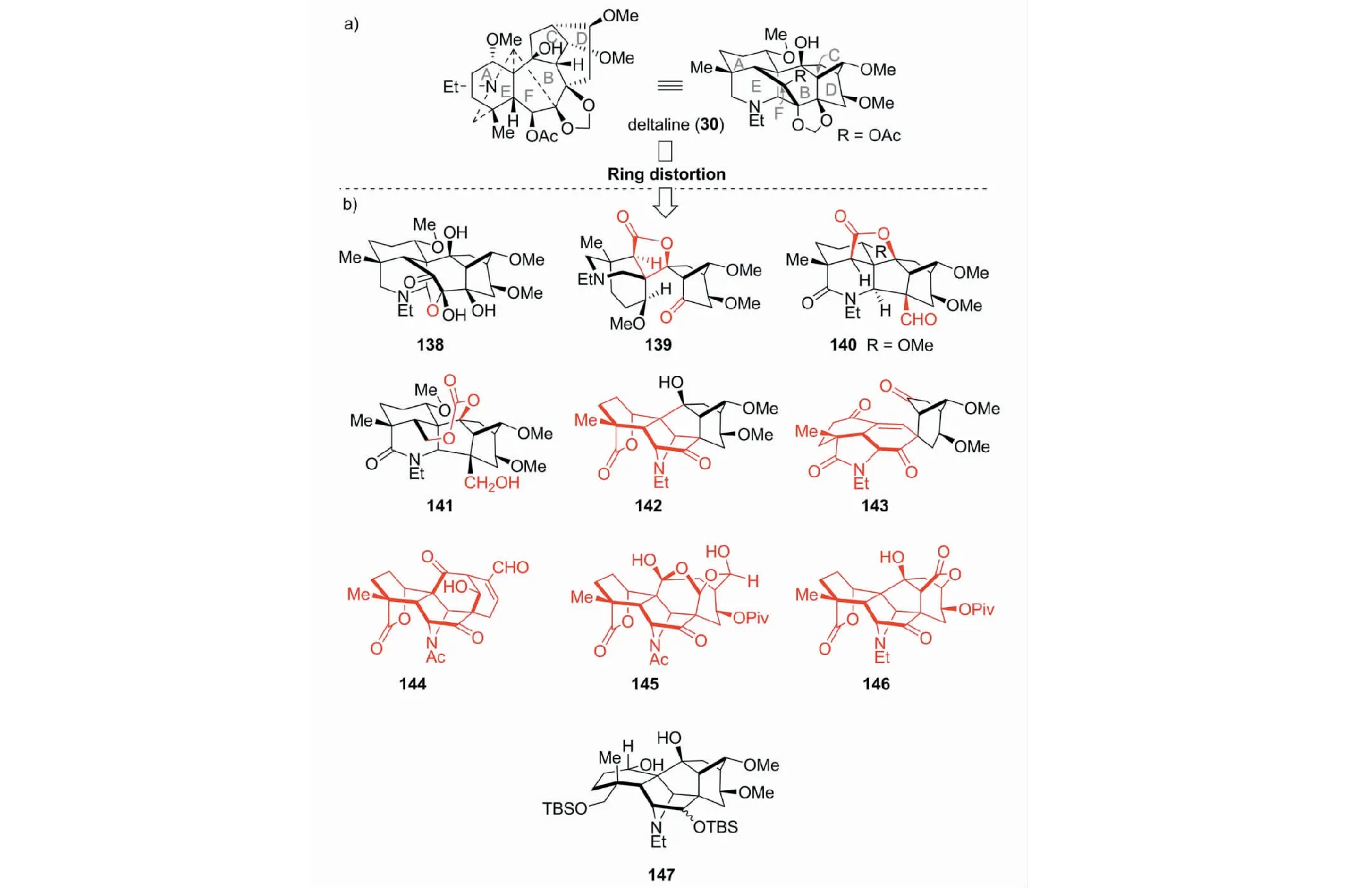
图32 基于二萜生物碱deltaline(30)多样性合成的代表性化合物Fig.32 Representative products from diterpenoid alkaloid deltaline (30)
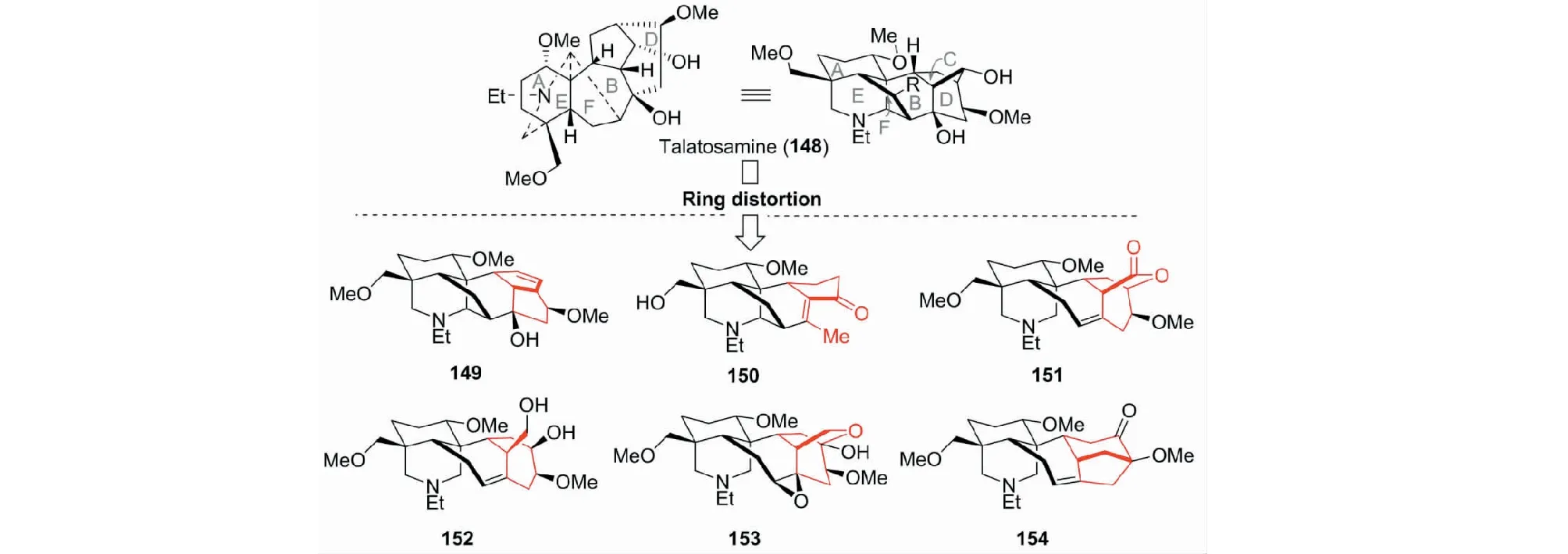
图33 基于二萜生物碱talasamine(148)多样性合成的代表性化合物Fig.33 Representative products from diterpenoid alkaloid talasamine (148)
7 附子中强心活性成分及其构效关系的研究
附子是著名中药,广泛用于临床。其强心作用研究成为数十年来国内外研究的热点与难点。文献报告的强心活性成分,如dl-去甲乌药碱、尿嘧啶和附子苷等含量甚微,难于被认可为附子中强心的主要成分。我们采用体外牛蛙离体心脏模型追踪筛选,首次从其水溶性活性部位,分出具有显著强心作用的二萜生物碱N-去乙基乌头宁碱(N-deethylaconine,155)、北乌宁碱(beiwutinine,156)、次乌宁碱(hypaconine,157)和中乌宁碱(mesaconine,158),以及具短暂强心作用的附子灵(fuziline,159)[56,57]。
同时,设计、制备数十个化合物,供强心活性筛选,并首先报告其构效关系:N甲基或NH、8-OH基和15α-OH基,以及胺醇形式是强心活性的必需结构(见图34A)[19,58]。最后,通过体内试验(结扎大鼠冠状动脉左前降支致心衰,戊巴比妥钠致心衰大鼠模型,以及阿霉素致小鼠慢性心衰模型),发现一种具有高效(有效剂量:0.0375 mg/kg,0.1 mol/kg,大鼠,iv;5 mg/kg:小鼠灌胃)、低毒(LD50:643.37 mg/kg,ip,小鼠;135.04 mg/kg,iv,小鼠),新的作用机制的原料易得,制备简便的新类型的侯选强心药物中乌宁碱[57,59]。目前正在进行临床前研究。所有这些,都是附子强心作用研究的新突破。
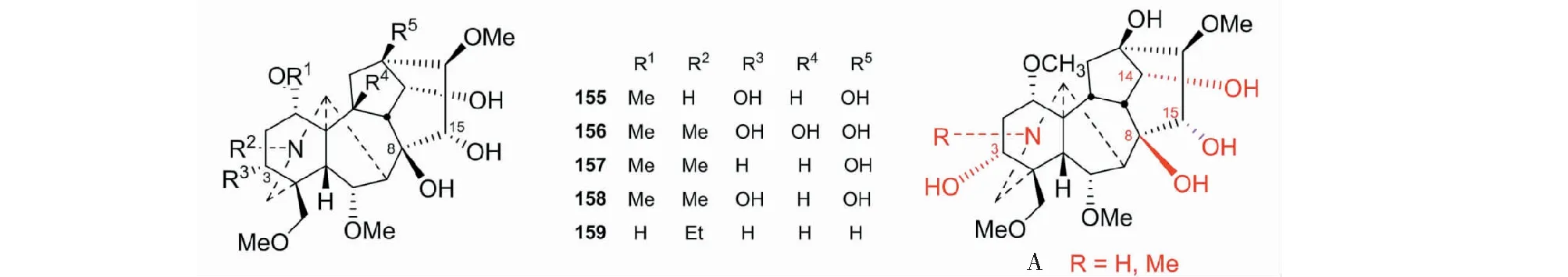
图34 具有强心作用的二萜生物碱155~159及其构效关系(A)Fig.34 Diterpene alkaloids 155-159 with cardiotonic effect and their structure-activity relationships(A)
8 小结
回顾我们的二萜生物碱化学的研究历程,最令人难忘的是艰难而持久进行的由乌头碱类转化合成紫杉烷类似物的研究,以及全合成和附子中抗心衰中乌宁碱的发现、研究与开发。
纵观二萜生物碱的研究,除了继续深入地开展植物化学的研究外,对其多方面的生物活性做深入的研究,以及世界范围内天然产物化学家对二萜生物碱全合成的继续关注,将成为未来二萜生物碱化学研究的新趋势。
致谢:衷心地感谢我的导师们梁晓天院士、方起程研究员、于德泉院士和S.W.Pelletier教授的培养与教诲!还应该感谢我的同事、研究生和博士后们的支持与努力!他/她们的名字都列在本文的参考文献中。本院天然药物系刘小宇副教授和唐培副教授协助加工与排版,特此致谢!
