离子色谱法同时测定环境空气中氨、甲胺、二甲胺和三甲胺
2021-08-24牛爽潘素素商宇扬霍胜伟张国城杨振琪
牛爽,潘素素,商宇扬,霍胜伟,张国城,杨振琪
(北京市计量检测科学研究院,国家生态环境监测治理产品质量监督检验中心,北京 100029)
氨在常温常压下是一种无色而具有强烈刺激 性恶臭的气体。胺是氨分子中一个或多个氢原子被烃基取代后的衍生物,甲胺、二甲胺、三甲胺具有氨味或鱼腥味。氨和有机胺类物质是仅次于有机硫化合物的恶臭污染物[1]。氨和有机胺是重要的化工原料,广泛地应用于多种行业。氨和大部分有机胺具有刺激性和毒性,对眼睛,耳、鼻、喉,皮肤,呼吸道,肝脏和肾脏有损伤,还可以与亚硝酸盐反应,生成强致癌性的亚硝胺类物质,危害人体健康[2–3]。有研究报道胺可促进新粒子形成及亚微粒子增长,并参与二次有机气溶胶的形成,直接或间接影响环境空气质量和区域气候变化[4–5]。
GB/T 18883—2002规定室内氨的浓度在1 h内的均值不大于0.2 mg/m3[6]。GBZ 2.1—2019规定工作场所空气中氨的时间加权平均容许浓度(PC–TWA)为20 mg/m3,短时间接触容许浓度(PC–STEL)为30 mg/m3;甲胺、二甲胺的PC–TWA和PC–STEL分别为5、10 mg/m3[7];氨和三甲胺作为恶臭污染物,GB 14554—1993对无组织排放源的限值分别为1.0、0.05 mg/m3(一级)[1]。因此环境空气中氨和有机胺的准确测定对污染源控制和环境保护具有重要意义。
目前,空气中氨的检测方法国家标准GB/T 18204.25—2000[8]、GB/T 14668—93[9]、GB/T 14679—93[10]和HJ 533—2009[11]、HJ 534—2009[12]均采用分光光度法;GB/T 14669—93[13]采用氨气敏电极法,由测得的电位值确定吸收液中氨的含量;GB/T 14676—93[14]和GBZ/T 160.69—2004[15]均采用气相色谱法分别检测恶臭污染源排气、厂界环境空气和工作场所空气中的三甲胺。
氨的标准分析方法有分光光度法、离子选择电极法和离子色谱法。离子选择电极法和分光光度法灵敏度不高,且无法实现氨、甲胺、二甲胺和三甲胺的同时测定。国内现行的检测环境空气中的三甲胺的标准方法只有气相色谱法,GB/T 14676—93采用经过草酸处理的玻璃微球吸附,通过向采样管中注入饱和氢氧化钾溶液和氨气,使采集的三甲胺游离成气态并进入经真空处理的解吸瓶后进行气相色谱分析。气相色谱法可以实现氨、甲胺、二甲胺和三甲胺的同时测定,但是样品处理步骤比较繁琐,分析成本较高。
冯顺卿[16]等以稀盐酸作为吸收液采集大气中的三甲胺,用离子色谱法进行测定,环境空气采样体积为10 L、吸收液体积为100 mL时,环境空气检出限为1.0 mg/m3。郑波[17]采用经草酸处理的玻璃纤维滤膜采集大气中的氨及低级脂肪胺,用离子色谱法进行分析,采样体积为2.4 m3时,氨与三甲胺的检出限分别为2×10–4、2×10–3mg/m3。龚文杰[18]等以稀硫酸为吸收液,用非抑制型电导检测器离子色谱法检测,乙醇胺和三甲胺在质量浓度为0~200 μg/mL的范围内相关系数均大于0.999,检出限分别为1.9、2.4 μg/mL。邓迈华[19]用稀硫酸为吸收液,以10 mL吸收液采气60 L,进样体积为100 μL,氨、二甲胺、三甲胺最低检出质量浓度分别为0.017、0.017、0.133 mg/m3。研究表明离子色谱法检测氨和低级脂肪胺,可实现目标物的同时测定,采样简单,方法快速、灵敏、准确[16–20]。2019年12月生态环境部发布了HJ 1076—2019 《环境空气氨、甲胺、二甲胺和三甲胺的测定 离子色谱法》[21],样品经滤膜过滤,被稀硫酸吸收液吸收后,经阳离子色谱柱交换分离,采用离子色谱电导检测器进行检测。
笔者参考HJ 1076—2019,采用离子色谱法同时测定环境空气中的氨、甲胺、二甲胺和三甲胺,发现三甲胺的色谱保留时间受其浓度影响,探讨了色谱保留时间漂移的规律。
1 实验部分
1.1 主要仪器与试剂
离子色谱仪:ICS–1100型,美国戴安公司。
大气采样器:KB–6E型,青岛精诚仪器仪表有限公司。
多孔玻板吸收管:四川蜀玻集团有限责任公司。
氯化铵标准物质:编号为GBW(E) 060322,质量分数为99.96%,相对扩展不确定度为0.08%(k=2),国防科技应用化学一级计量站。
甲胺盐酸盐:质量分数为98.0%,美国阿法埃莎化学有限公司。
盐酸二甲胺:质量分数为99.0%,北京伊诺凯科技有限公司。
三甲胺盐酸盐:质量分数为98.0%,北京伊诺凯科技有限公司。
硫酸溶液:c(1/2 H2SO4)=0.100 4 mol/L,北京海岸鸿蒙标准物质技术有限责任公司。
甲磺酸:质量分数为98%,北京伊诺凯科技有限公司。
标准储备液:分别称取0.392 8 g氯化铵、0.277 2 g甲胺盐酸盐、0.228 4 g盐酸二甲胺、0.206 3 g三甲胺盐酸盐,分别用水溶解后于250 mL容量瓶中定容。质量浓度均为500 mg/L。
硫酸吸收液:c(1/2 H2SO4)=0.01 mol/L,现用现配。
实验用水为新制备的无氨水。
1.2 仪器工作条件
阳离子分析柱:IonPac CS16柱(250 mm×5 mm,美国戴安公司);阳离子保护柱:IonPac CG16柱(50 mm ×5 mm,美国戴安公司);检测器:抑制型电导检测器,抑制电流为65 mA;淋洗液:22 mmol/L甲磺酸溶液,流量为1.0 mL/min;进样体积:25 μL。
1.3 实验步骤
1.3.1 样品处理
取10.0 mL硫酸吸收液装入多孔玻板吸收管中,用硅胶管和特氟龙管连接至空气采样器,以0.5 L/min的流量采集环境空气样品60 min。样品采集后,用硅胶管封闭多孔玻板吸收管的进气口和出气口,直立运输和保存。
用适量水淋洗吸收管,将样品全量转入10 mL具塞比色管中,定容至标线,摇匀。用微孔滤膜过滤,弃去2 mL初滤液,收集适量续滤液待测。
1.3.2 标准曲线的制作
将标准储备液配制成混合标准使用液,氨质量浓度梯度为0.025、0.05、0.25、1.00、5.00 mg/L,有机胺质量浓度梯度为0.10、0.20、1.00、4.00、20.0 mg/L,进样测定,记录保留时间和色谱峰面积,每种浓度的溶液重复进样5次,以色谱峰面积平均值为纵坐标、对应的化合物质量浓度为横坐标进行线性回归,建立标准曲线线性方程。
1.3.3 质量控制与质量保证
每批样品做一个实验室空白和全程序空白,色谱条件与样品测定保持一致。结果显示全程序空白和实验室空白测定结果一致,均未检出目标物,因此在测定实际样品时不需扣除背景值。在考察线性关系时,采用质控样品进行验证,相对误差控制在10%以内。分析实际样品时,做空白加标试验。
2 结果与讨论
2.1 氨、甲胺、二甲胺和三甲胺色谱图
采用抑制型电导检测器,甲磺酸作为淋洗液,用IonPac CS16阳离子色谱柱进行分离,得到氨、甲胺、二甲胺和三甲胺混合标准溶液的离子色谱图如图1所示。由图1可以看出,4种物质分离良好。氨(1.00 mg/L)、甲胺(4.00 mg/L)、二甲胺(4.00 mg/L)、三甲胺(4.00 mg/L)的色谱保留时间分别为12.011、13.331、16.517、25.784 min。

图1 氨、甲胺、二甲胺和三甲胺的标准离子色谱图
氨、甲胺、二甲胺的保留时间比较稳定,相同色谱条件下,受浓度影响较小。三甲胺的保留时间会随着样品浓度的不同而发生“位移”[17],如图2所示。标准溶液浓度为1.04、4.12、15.2、20.2 mg/L的三甲胺色谱保留时间分别为26.331、25.084、24.260、23.614 min,最大位移为2.717 min。三甲胺保留时间发生“位移”是有规律的,随着浓度的增加,保留时间会缩短。
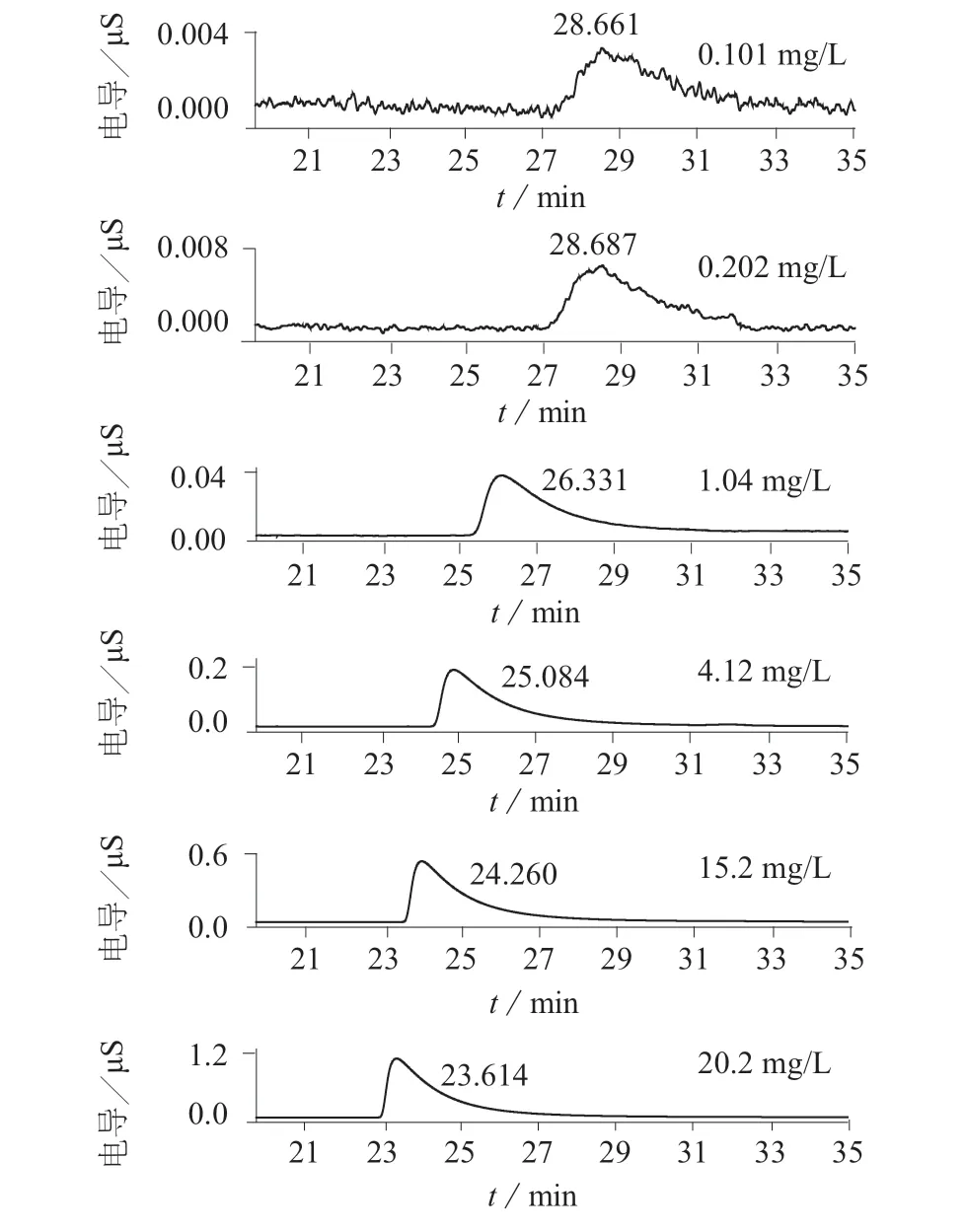
图2 不同浓度的三甲胺色谱图
为验证这种“位移”不同于保留时间的波动和漂移,对同一浓度(4.12 mg/L)的三甲胺进行分析,结果显示最大波动为0.027 min(图3)。因此,浓度相同,三甲胺的保留时间比较稳定;浓度不同,其保留时间存在“位移”现象。三甲胺的色谱峰常见拖尾呈“鲨鱼鳍”状,保留时间随着溶液浓度的增大而缩短。有研究认为这种现象可能是由色谱柱内质量过载造成的[23]。这种质量过载现象认为是带同种电荷的离子之间存在着相互排斥力,导致其在固定相表面过早发生非线性吸附[24]。为了验证这种说法,采用浓度更低的三甲胺溶液进行检测,质量浓度分别为0.101、0.202 mg/L,得到的保留时间分别为28.661、28.687 min。降低浓度,三甲胺的保留时间进一步增加,但是浓度低到一定程度时,保留时间波动不大,进一步说明三甲胺保留时间发生“位移”是由分析柱过载造成。虽然降低浓度或减小进样体积可以改善色谱柱质量过载,但浓度过低,色谱峰变小,将会引起显著的定量误差。三甲胺保留时间随浓度发生“位移”的情况,说明该色谱柱不适合于三甲胺的分离和检测,但是不影响使用,在色谱峰附近没有其它离子峰产生干扰时,可通过手动积分的方式,对三甲胺进行定量分析。

图3 4.12 mg/L三甲胺溶液色谱图
采用与混合标准溶液相同的分析方法,分析了加标的环境空气样品,氨、甲胺、二甲胺、三甲胺的加标质量浓度分别为0.10、0.17、0.17、0.33 mg/m3,得到的离子色谱图如图4所示。

图4 加标环境空气样品的离子色谱图
由图4可知,氨、甲胺、二甲胺、三甲胺的保留时间分别为11.914、13.257、16.497、25.357 min。从图4可以看出,4种组分分离良好,且基质干扰较少。
2.2 线性方程和检出限
按照1.3.2试验并进行线性回归,计算线性方程、相关系数。按照样品分析的全部步骤,空白试验中未检测出目标物质。依据HJ 168—2010[25],对浓度为估计方法检出限2~5倍的样品进行n(n≥7)次平行测定,并计算方法检出限。以4倍检出限作为测定下限。氨、甲胺、二甲胺和三甲胺的线性范围、线性方程、相关系数、检出限和测定下限列于表1。由表1可知,氨、甲胺、二甲胺和三甲胺的相关系数为0.999 3~1.000,当环境空气采样体积为30 L、吸收液体积为10 mL时,氨、甲胺、二甲胺和三甲胺的检出限分别为0.003、0.007、0.003,0.004 mg/m3。能够满足定量分析的要求。

表1 氨、甲胺、二甲胺、三甲胺的线性方程、线性范围、相关系数、方法检出限及测定下限
2.3 精密度试验
对某空气样品进行6次重复测定,测定结果列于表2。

表2 精密度试验结果
由表2可知,氨、甲胺、二甲胺、三甲胺测定结果的相对标准偏差分别为1.7%、0.8%、1.7%、1.2%,表明本法精密度良好。
2.4 加标回收试验
对含有目标化合物中、低两种浓度的实际空气样品进行加标回收试验,氨、甲胺、二甲胺、三甲胺的加标质量浓度分别为0.10、0.17、0.17、0.33 mg/m3和0.02、0.03、0.03、0.17 mg/m3,试验结果列于表3。

表3 加标回收试验结果
由表3可知,氨、甲胺、二甲胺、三甲胺的加标回收率为96.0%~115%,测量准确度满足分析要求。
2.5 比对试验
采用本方法测定了国家二级标准物质氯化铵[氯化铵的质量分数为99.96%,相对扩展不确定度为0.08%(k=2)],配制溶液质量浓度为2.5 mg/L,测定结果为2.479 4 mg/L,相对误差为0.8%,进一步验证了本方法的准确性。
3 结语
参考HJ 1076—2019,采用离子色谱法同时测定环境空气中的氨、甲胺、二甲胺和三甲胺,以硫酸溶液作为吸收液,对空气样品进行采样,氨、甲胺、二甲胺和三甲胺的离子色谱峰分离良好,并且无其它明显的阳离子干扰测定。三甲胺常见拖尾现象,而且随着浓度的增大,保留时间会缩短,发生“位移”;浓度很低时,保留时间没有显著差别,这可能是色谱柱“过载”引起。氨、甲胺、二甲胺、三甲胺的色谱峰面积与质量浓度线性关系良好,分析精密度和准确度能够满足实际环境空气样品检测的需求。
