基质细胞在炎症性肠病肠道免疫中的作用
2021-08-12杨理超吴国涛袁联文
杨理超,吴国涛,袁联文
(中南大学湘雅二医院老年外科,湖南长沙 410011)
[提要] 炎症性肠病(inflammatory bowel disease)是一种发病率持续上升的慢性自身免疫性疾病,它可以累及全消化道,它的疾病表现形式多种多样(如腹痛、腹泻、梗阻等)。炎症性肠病的发病机制尚未完全清楚,所以对于它的治疗暂未有突破性的进展。但可以明确的是肠道免疫在IBD的发病过程发挥着必不可少的作用,近年来诸多研究表明多种基质细胞能与人体的免疫细胞相互影响,从而促进或者抑制IBD的炎症发展。本文就基质细胞在IBD的肠道免疫中的作用进行综述。
1 引 言
在哺乳动物的胃肠道中栖息着数百种不同种类的共生微生物,它们与宿主之间存在着一种互惠互利的关系。同时,某些可导致组织损伤的病原微生物也可能在肠腔聚集。因此,肠道的免疫系统扮演着极其重要的角色,它既需要对致病的病原菌做出快速且有效的免疫反应,还同时需要对食物和“肠道益生菌”有着良好的耐受性。一旦这种微妙的平衡出现紊乱就可能会导致肠道内出现异常的炎症反应,导致慢性肠炎,如炎症性肠病(inflammatory bowel disease, IBD)[1]。IBD是一种免疫介导的炎症性疾病,可引起胃、肠道乃至全身细胞的炎症,在世界范围内呈不断上升的趋势。虽然IBD的发病机制尚未明确,但目前已知引发和维持炎症的因素很多,包括先天性和获得性免疫因素、遗传因素、胃肠道微生物群的改变和某些环境因素等[2]。目前关于IBD的研究主要集中在胃肠道的免疫细胞方面,大多数学者认为免疫细胞属于基质细胞的一部分,而最新的研究发现其他的基质细胞在IBD发病机制中也有着重要作用[3]。本文旨在回顾IBD中的先天性和获得性免疫反应,以及描述其他肠基质细胞在这些反应中所扮演的角色。
2 IBD中的免疫细胞
在肠道中共同存在着先天性免疫和获得性免疫,它们相辅相成,共同维护着肠道稳态。先天肠道免疫系统由肠粘膜上皮屏障、天然免疫细胞、先天免疫分子等组成。先天免疫可以看作是启动获得性免疫的基石,为原始T细胞提供抗原和激活信号,它还参与获得性免疫中免疫细胞的成熟、分化和归巢,从而调节机体免疫功能[4]。先天性免疫细胞包括了树突状细胞(dendritic cell, DC)、巨噬细胞(macrophages)、先天性淋巴细胞(innate lymphoid cells, ILCs)等。DC是一种特殊的抗原提呈细胞(antigen presenting cell, APC),负责活化T细胞和诱导获得性免疫反应,在先天性免疫和获得性免疫之间扮演着关键角色[1]。DC由两个主要亚群组成:常规树突状细胞(conventional DCs, cDC)和浆细胞样树突状细胞(plasmacytoid DCs,pDC)。巨噬细胞是一种具有强力吞噬功能的细胞,可以清除细菌和细胞碎片,这对维持组织内环境稳定至关重要。胃肠道的大部分巨噬细胞主要是由血源性单核细胞通过一系列中间体来补充的,但肠道环境因素对其独有的“免疫调节”的影响仍不完全清楚[5]。肠道巨噬细胞主要通过产生白介素10(interleukin-10, IL-10)来促进调节性T细胞(regulatory cells,Treg)的增殖,从而防止机体对无害的共生微生物产生过度的炎症反应。同时肠巨噬细胞还产生多种细胞因子和可溶性介质来维持肠道稳态,如前列腺素E2(prostaglandin E2,PGE2)、骨形态发生蛋白2(bone morphogenetic protein 2,BMP2)[6]。ILCs根据其表型和功能的异质性可分为三组,其中第一组ILCs的特征是在IL-12和(/或)IL-15作用下产生辅助性T1( T helper 1,Th1)细胞相关的干扰素-γ(interferon-gamma, IFN-γ)和肿瘤坏死因子(tumor necrosis factor, TNF),其包括经典的自然杀伤(natural killer, NK)细胞和先天淋巴细胞亚群1(innate lymphoid cells 1, ILC1)细胞[7]。第二组ILC(ILC2s)产生与Th2细胞相关的细胞因子IL-5和IL-13,它的增殖和激活主要靠由上皮细胞来源的细胞因子IL-25和IL-33等[8]。第三组ILC在IL-23刺激下产生TH17和TH22细胞相关细胞因子,包括IL-17和IL-22[7]。与先天免疫反应相比,获得性免疫系统具有高度的特异性。获得性免疫的主要参与者是T淋巴细胞,启动获得性免疫反应的关键步骤包括激活Th0淋巴细胞(Th1、Th2和Th17细胞)和抑制Treg细胞的活性[9]。Th1细胞参与细胞介导的炎症和迟发型超敏反应,对消除细胞内病原体有着非常重要的作用[10]。以前常常把Th1细胞定义为产生IL-2和IFN-γ的免疫细胞,但研究表明Th1细胞还可以产生许多其他的细胞因子,如TNF、粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony stimulating factor, GM-CSF)等[11]。Th2细胞因其在对抗寄生虫感染以及参与过敏性疾病中的作用而被人们所认识,它主要在上皮组织中发挥作用,尤其是肠道和肺部,活化后主要产生IL-13和IL-5[12]。Th17途径通过激活信号转导子和转录激活因子3(signal transducers and activators of transcription 3,Stat3)通路并导致IL-17和IL-21的分泌[2]。Th17细胞分泌的IL-17有很多作用,包括:中性粒细胞的募集,天然免疫细胞的激活,增强B细胞的功能,以及诱导包括TNF、GM-CSF和IL-1β在内的促炎细胞因子的释放。此外,IL-17还可诱导基质金属蛋白酶(matrix metalloproteinase, MMPs)与抗菌肽的产生来促进组织损伤[13]。Treg细胞发育依赖于叉头框蛋白P3(forkhead box protein P3, FOXP3)转录因子的表达,缺乏FOXP3可导致机体出现致死性自身免疫性疾病[14]。Treg介导的免疫抑制的机制包括抗炎细胞因子的分泌和抑制性受体的表达[15]。而与Treg最相关的两种细胞因子是IL-10和TGF-β。研究发现TGF-β和FOXP3之间存在正反馈,其在维持外周免疫耐受方面起着关键作用[16]。
3 免疫细胞和基质细胞的交互作用
免疫细胞和其他基质细胞之间的相互作用对伤口愈合、癌症和广泛的炎症性疾病至关重要。在人的肠壁各层都能检测到基质细胞。通常来说,最丰富的基质细胞是成纤维细胞,其次是肌成纤维细胞、平滑肌细胞、周细胞和间充质基质细胞等[3]。众所周知,肠系膜脂肪组织(mesenteric adipose tissue, MAT)的增生是克罗恩病(Crohn disease,CD)的特征。而正是来源于MAT的基质细胞可以释放脂肪因子和细胞因子,在导致肠道炎症的病理机制中起着极其重要的作用[17]。越来越多的学者关注基质细胞在协调炎症反应中的作用(图1),因为基质细胞与免疫细胞之间的相互影响似乎可以调控炎症的走向,这为治疗炎症性疾病开拓了新的视野。
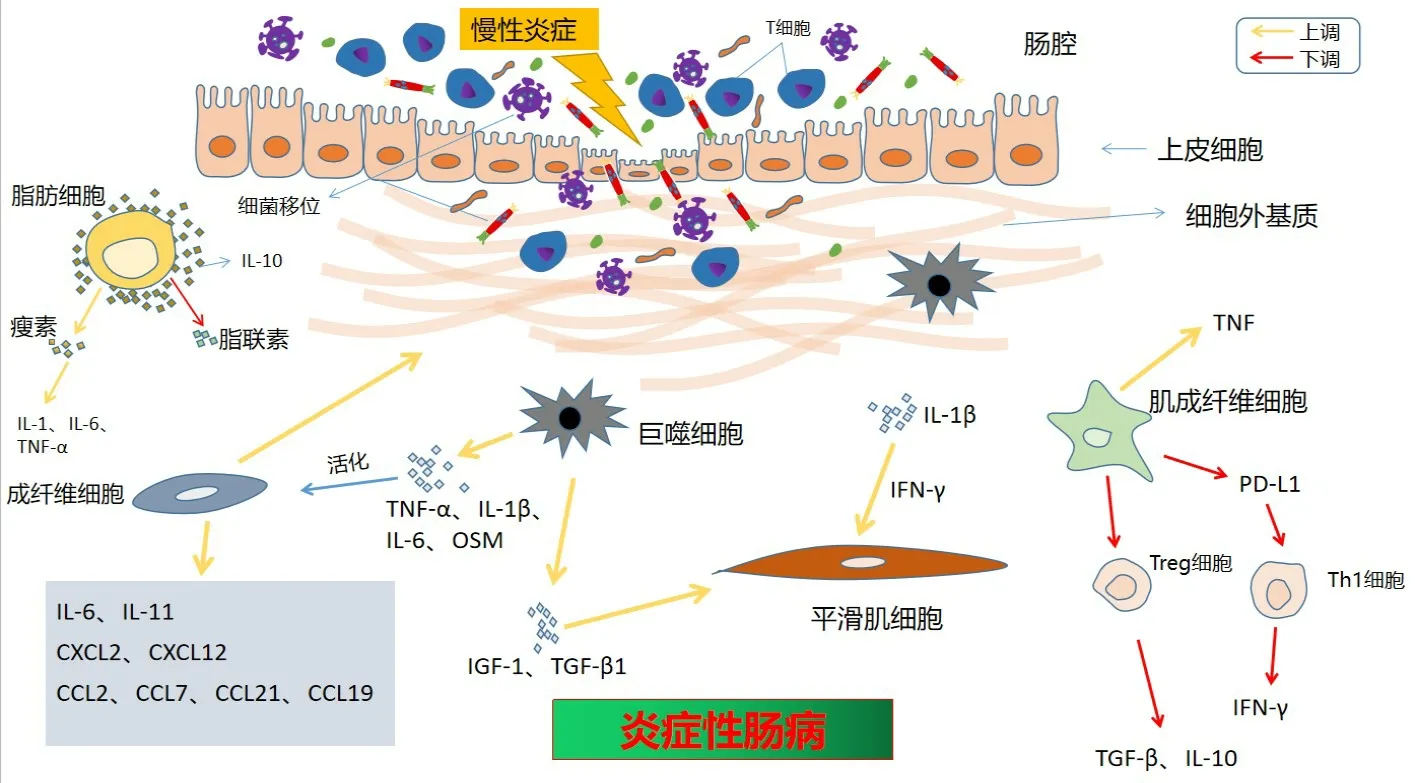
图1 多种基质细胞在IBD肠道免疫应答中的相互作用
3.1 肠成纤维细胞
众所周知,肠成纤维细胞过度产生的细胞外基质(extracellular matrix, ECM)会使得IBD患者发生肠道纤维化。它主要通过分泌Ⅰ型、Ⅱ型和Ⅴ型胶原和纤维连接蛋白,以及通过包括MMPs在内的蛋白水解酶重塑基质,从而在ECM的产生中发挥作用[18]。为了修复在慢性炎症(如IBD)中受损的肠道上皮,成纤维细胞的迁移和胶原的沉积在上皮重建中发挥必不可少的作用。但体外研究显示,IBD患者的肠成纤维细胞的迁移能力较对照组降低,CD瘘管患者的成纤维细胞的迁移能力甚至进一步降低,这可能是CD瘘管和纤维化形成的原因[19]。虽然IBD患者肠纤维细胞的增殖能力有所降低,但其在急性期时的增殖能力却大幅增加,同时产生更多的胶原[20]。糖蛋白抗人平足蛋白(podoplanin, PDPN)的高表达与肠成纤维细胞有关,而PDPN是一种已被证实在CD或溃疡性结肠炎(ulcerative colitis, UC)患者的病变组织中大量存在的标志[3]。在IBD的肠道中,成纤维细胞可以产生单核细胞趋化因子(如CCL2,CCL7)、T细胞募集因子(如CXCL2,CCL19,CCL21,和CXCL12)、中性粒细胞引诱剂(如CXCL2,CXCL8,和CXCL1),和参与纤维化的因子(如IL-11)[21-22]。抑瘤素M(oncostatin M, OSM)由造血细胞产生,通过抑制骨髓基质细胞向脂肪细胞的分化而调节骨髓基质细胞[23]。OSM是治疗IBD的潜在生物标记物和治疗靶点,对抗TNF耐药患者具有特殊的相关性,OSM mRNA在CD和UC肠粘膜组织中的表达显著增加,OSM/OSMR的表达与IBD的组织病理学严重程度密切相关[24-25]。在UC结肠炎症期间,结肠成纤维细胞显示出OSMR的高表达[21],通过产生各种促炎分子(如IL-6、白细胞粘附因子)来响应OSM[24]。除OSMR外,肠成纤维细胞还表达IL-17受体。CD患者的肠道免疫细胞产生IL-17增多,可作用于肠成纤维细胞,诱导转录因子NFKBIZ和促炎趋化因子CXCL1的表达[26]。这可能会影响成纤维细胞的活性。IBD中的巨噬细胞还可以产生炎性细胞因子(如TNF-α、IL-1β、OSM和IL-6)来影响成纤维细胞的活化[22,24]。
3.2 肌成纤维细胞
肌成纤维细胞的激活与组织损伤和炎症有关。它们能够迁移到受损部位,在那里它们收缩伤口区域,并产生细胞外基质成分,恢复和重塑受损的粘膜组织[27]。而在炎症环境中肌成纤维细胞的持续激活可能会造成受损组织的不可逆损害,因为它可以促进粘膜下层的纤维化和相应细胞的增殖[28]。结肠肌成纤维细胞(colonic myofibroblasts, CMFs)是一种新型的非专业性抗原提呈细胞,在正常人结肠粘膜中含量丰富。CMFs通过改变基质沉积和促进促炎细胞因子的产生,积极参与IBD相关炎症的进展[29]。研究发现正常粘膜来源的CMF通过维持FOXP3的表达来刺激Treg细胞的增殖,而且增殖的Treg通过表达中等水平的IL-10和高水平的TGF-β1来介导其免疫抑制作用。而IBD来源的CMF诱导活化Treg细胞能力减弱,反而产生一种CD127阳性的T细胞亚群,这可能意味着TGF-β1和IL-10的表达减少[30]。与健康对照组的结肠相比,CD急性期的肠肌成纤维细胞的程序性细胞死亡配体PD-L1的表达显著减少,而PD-L1表达的减少可能导致Th1细胞产生IFN-γ的减少[31]。IBD患者的肠肌成纤维细胞比来自健康对照组的非炎症细胞或肠肌成纤维细胞表达更多的TNF,这为抗TNF治疗提供了一条新的途径。
3.3 平滑肌细胞(smooth muscle cells, SMC)
肠纤维化是IBD患者的常见症状,30%以上的CD患者和5%左右的UC患者会出现这种症状。在CD相关的“纤维狭窄”中,平滑肌增生或肥大对肠道狭窄的作用最大,而对纤维化的影响可能较小[28]。TNF-α与平滑肌上的TNF-R1和R2结合,导致核因子-kB(nuclear factor kappa B, NF-kB)的激活和许多趋化基因的表达,尤其是单核细胞趋化蛋白-1(monocyte chemotacic protein-1, MCP-1)、IL-8和细胞内粘附分子-1(intercellulor cell adhesion molecule-1, ICAM-1)[32]。Th17细胞产生的细胞因子(如IL-17、IL-17RA和IL-17RB等)的受体也能在肠道平滑肌上找到[33]。除了巨噬细胞外,肌间神经丛和平滑肌细胞周围几乎没有找到其它炎症细胞。而巨噬细胞产生胰岛素样生长因子-1(insulin-like growth factor-1, IGF-1)和TGF-β1在平滑肌肥大和增生中起重要作用,其中IGF-1通过刺激细胞增殖和抑制细胞凋亡来调节肠平滑肌生长[34]。有趣的是,尽管Th1和Th2途径产生的细胞因子会导致平滑肌肥大和增生,但它们对平滑肌的收缩性却有相反的影响[33]。
3.4 肠系膜脂肪组织(mesenteric adipose tissue, MAT)
在CD患者的病变回肠中可以观察到病变区域的淋巴结和肠道周围脂肪组织的选择性增大,增大的脂肪组织能够覆盖超过一半的肠道表面,而这就是肠系膜脂肪,也称为爬行脂肪。它是导致IBD患者TNF-α、IL-6和其他促炎因子升高的常见原因,同时可释放大量脂肪因子来加速或抑制炎症的进程[35]。爬行脂肪相对于其他的内脏脂肪而言更具有免疫活性,它的丰富程度与病变组织的炎症程度密切相关(如巨噬细胞的浸润程度)[36]。CD患者的MAT中有大量的M2亚型巨噬细胞浸润并释放大量细胞因子,构成了一个富含IL-10的微环境,有可能限制肠道炎症的发展[37]。而浸润在脂肪组织中的巨噬细胞不仅是许多细胞因子的来源,也可在某种程度上调节脂肪细胞的分泌活动。有研究发现来源于脂肪细胞的脂肪因子(如瘦素)可通过增加巨噬细胞对T细胞的趋化能力来增强其抗炎作用[37],有意思的是,虽然CD患者的肠系膜脂肪中浸润大量的抗炎性巨噬细胞,但在肠道固有层却会聚集大量促炎性巨噬细胞,而这些促炎性巨噬细胞通过破坏肠上皮紧密连接蛋白的调节,诱导上皮细胞凋亡,直接削弱肠上皮屏障,从而促进肠炎的发生[38]。瘦素是主要由白色脂肪组织分泌的一种脂肪因子,它最重要的功能是调节人体内能量平衡和代谢水平,如向下丘脑传递信号来调节食欲。瘦素在IBD中具有很强的促炎作用,它可在炎症刺激下释放大量细胞因子(如IL-1、IL-6)[39]。在DSS引起的结肠炎小鼠模型中,抑制瘦素的产生可减缓炎症的发展,因此瘦素可被认为是一种促炎性脂肪因子,所以减少瘦素的产生也许是IBD的潜在治疗方法[40]。而与瘦素不同,脂联素(adiponectin, APN)是一种较为丰富的来源于脂肪细胞的抑炎性脂肪因子。有研究[41]表明,与正常回肠远端相比,活动性回肠CD患者的黏膜脂联素表达降低。该脂联素被证明具有抑制粘附因子、金属蛋白酶(MMP9-13)和促炎介质表达的功能,可减缓IBD疾病的发展进程[42]。有研究[43]表明替米沙坦可改善IBD,其之所以能够减少内脏脂肪正是通过减少瘦素的产生并增加脂肪组织中APN的表达。在CD患者中,其疾病的跨壁性炎症的特征大大增加了细菌向爬行脂肪转移的机会,肠系膜脂肪中的脂肪细胞可通过捕捉炎性信号,包括病原相关分子模式(pathogen-associated molecular patterns,PAMPs),模式识别受体如Toll样受体(Toll-like receptors, TLRs),以及核苷酸寡聚结构域受体1(nucleotide bing oligomerization domain 1, NOD1),从而对移位的肠道细菌产生反应[44],例如,脂肪细胞及其前体会因为脂多糖激活了NF-κB途径而导致TLR-4表达的增加,从而导致多种细胞因子和趋化因子(包括IL-6和TNF-α等)的产生增加。
在慢性炎症的刺激下,肠道上皮屏障不断减弱,肠道内细菌逐渐移位,而MAT中的脂肪组织呈炎性改变,释放大量细胞因子和脂肪因子(如瘦素),而抑炎性脂肪因子脂联素的表达下降,而且其周围浸润的巨噬细胞也会趋化T细胞来参与炎症反应。同时成纤维细胞不断产生大量细胞外基质,这是IBD患者最常见的并发症----肠梗阻发生的原因之一。肌成纤维细胞和平滑肌细胞也可产生大量的细胞因子来参与肠道炎症的发展。
4 结 语
虽然基质细胞在IBD发病机制中的作用尚未完全明确,但目前的研究已经显示肠道免疫细胞和不同亚群的基质细胞在炎症发展过程中具有相当大的作用。然而对于IBD基质细胞表达或刺激产生的许多细胞因子来说,它们在促进或抑制正在进行的炎症反应中的具体机制尚不完全清楚。直接针对在免疫细胞募集和激活中发挥作用的致病基质细胞的治疗方法有潜在的损伤全身基质细胞的风险,而且具体的致病基质细胞亚型还未最终确定。是否能像“粪菌移植(fecal microbiota transplantation, FMT)”[45]和“间充质干细胞移植”[46]来治疗IBD一样,通过引入“健康”或“抑炎性”基质细胞来抑制炎症免疫反应?这在未来需要进一步研究来证明这种治疗方法的可行性。
