1例表现为亚急性起病伴肌张力下降等不典型症状的肝豆状核变性
2021-08-05郑惠敏毛澄源史长河范丽媛骆海洋胡正威许予明
郑惠敏, 毛澄源, 秦 洁, 史长河, 范丽媛, 王 台 骆海洋, 张 槊, 胡正威, 范 雨, 杨 靖, 许予明
肝豆状核变性(hepatolenticular degeneration,HLD,0MIM277900)又称威尔逊病(Wilsons disease,WD),是一种由于ATP7B基因突变所导致的一种铜代谢异常的常染色体隐性遗传病,突变后基因编码的酶功能缺乏或者减弱,导致血清铜蓝蛋白(ceruloplasmin,CP)合成减少以及胆道排铜障碍,游离铜增多,常沉积于肝脏、神经系统、肾脏、角膜等部位,引起进行性加重的肝脏损伤、锥体外系症状、精神症状、肾损伤及角膜K-F环的出现[1]。患者常于青少年时期发病,起病隐匿、临床表现复杂且不典型,极易造成漏诊和误诊,甚至影响预后[2,3]。该种疾病常为慢性起病,亚急性起病尤为少见,而神经系统受累常表现为肌张力增高,肌张力下降极为罕见。本研究报道了就诊于我院的1例以亚急性起病、并伴肌张力下降等不典型表现的肝豆状核变性患者,并阐述和讨论了肝豆状核变性的临床诊断相关要点及可能机制。
1 临床资料
患者,男,16岁,于2019年11月8日,以“双下肢行走不稳半月余,加重伴言语不清4 d”为主诉就诊我院。半月前情绪波动后出现双下肢行走不稳,未就诊,自患病以来上述症状进行性加重。4 d前无明显诱因出现行走不能,伴言语不清,无意识丧失,无头痛头晕,无恶心呕吐、饮水呛咳、吞咽困难,无肢体麻木、四肢抽搐等症状。于当地医院查头部CT,示双侧基底节区对称片状低密度影,双侧脑室增宽,性质待定,建议进一步行核磁共振检查。余检查未见异常,给予对症支持治疗未见缓解。既往体健,无家族性遗传病史。先证者其父亲、母亲和2个姐姐健康状况良好,无与患者类似疾病。
神经系统体格检查:意识清楚,精神一般,语言不流利,记忆力、理解力、判断力、计算力、定向力正常,查体合作。双侧视野无缺损。双眼无上睑下垂,眼球各方向运动充分,无眼震、复视,双侧瞳孔等大等圆,直接、间接对光反射灵敏。双侧角膜反射存在,下颌反射阴性。双侧额纹对称,眼裂等大,鼻唇沟对称,无口角偏斜。伸舌居中,无舌肌萎缩。四肢肌张力降低,双上肢肌力Ⅴ级,双下肢肌力Ⅳ级,双侧指鼻试验欠稳准,快速轮替试验缓慢,双侧跟膝胫试验欠稳准,Romberg征不配合。双侧腱反射对称减弱,双侧肢体深、浅感觉正常,双侧Barbinski征阴性,自主神经系统检查未见明显异常,脑膜刺激征阴性,神经干牵拉实验阴性。
辅助检查:葡萄糖:5.37 mmol/L;乳酸:3.80 mmol/L; K+:4.24 mmol/L;血清同型半胱氨酸(Hcy):28.55 μmol/L;血沉:1 mm/h;心衰指标未见异常;尿粪常规、甲功、肝肾功、维生素和单项补体等未见异常。腹部超声:肝脏弥漫性实质不均匀改变,脾脏增大伴脾静脉增宽,颈部血管、甲状腺、心脏、泌尿系超声未见异常。头部MRI平扫示:双侧大脑脚、双侧基底节区和双侧丘脑异常对称性斑片状长T1短T2信号(见图1)。24 h动态血压、动态心电图监测未见异常。

图1 双侧基底节区可见异常的对称性斑片状长T1短T2信号(箭头示)
诊断:结合患者症状及体征分析,患者年轻男性,既往无特殊病史,亚急性起病,以双下肢行走不稳、言语不清为主要症状。定位:(1)锥体外系?(2)小脑?定性:(1)中毒代谢性? (2)感染性?进一步行相关检查,头部增强MRI,肌电图、脑脊液检验均未见异常。进一步综合患者年龄特点、影像学双侧基底节区异常信号、肝脏弥漫性改变,脾脏增大,考虑肝豆状核变性可能。眼部检查:K-F环(+)。血清铜蓝蛋白:铜蓝蛋白:4.648 mg/dl,24 h尿铜:59 μg/24 h(职业病所检测)。请消化内科会诊建议行上腹部CT及肝穿刺检查,患者拒绝。基因检测:目标序列捕获测序发现先证者存在ATP7B基因的c.2333G>T(8号外显子,p.R778L)和c.2828G>A(12号外显子,p.G943D)复合杂合突变(见图2)。此双杂合突变分别来自于其未患病的母亲和父亲,符合常染色体隐性遗传疾病致病规律。诊断:肝豆状核变性,ATP7B基因p.R778L与p.G943D复合杂合突变。
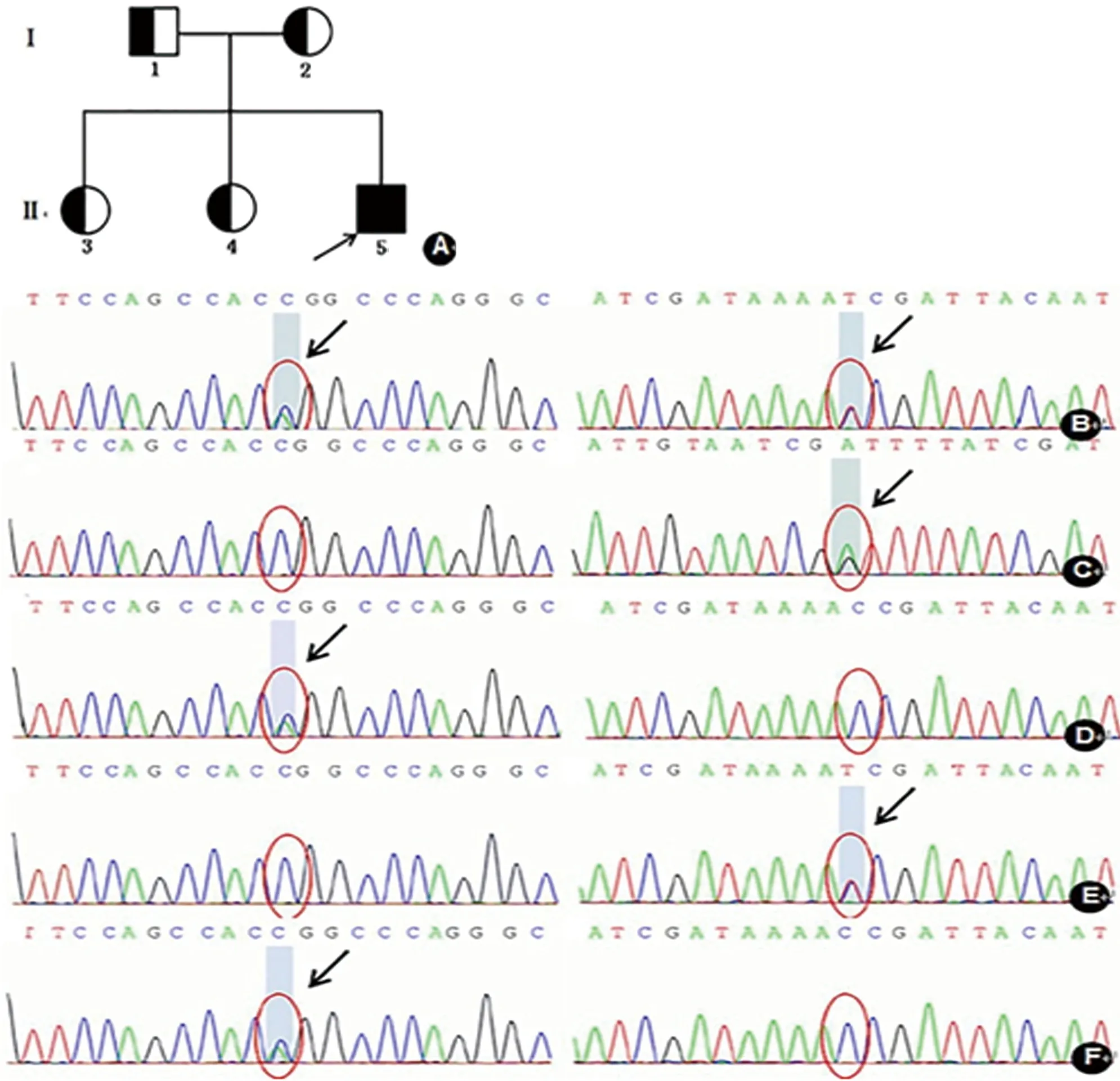
图2 家系资料和基因检测结果:家系图(A),ATP7B基因检测发现先证者8号外显子c.2333G>T和12号外显子 c.2828G>A突变(B),c.2333G>T来自其母(D),其二姐也为该位点的携带者(F),c.2828G>A来自其父(C),其大姐也携带了该突变位点(E),先证者为复合杂合突变。注:基因测序图中,除图E外,其余图示均为反义链。箭头示突变
治疗:嘱其低铜高蛋白饮食,并给予驱铜治疗(青霉胺片),加用降胱和营养神经治疗(甲钴胺片;维生素B1片;维生素B6片)。于2019年11月21日出院,给予青霉胺(根据病情及监测结果逐渐调整用量)、维生素B1片、维生素B6片和甲钴胺片。出院嘱患者低铜饮食,定期复查肝肾功能,24 h尿酮及铜蓝蛋白等指标。出院3 m后电话随访,患者自诉病情明显改善。
2 讨 论
肝豆状核变性(HLD)是一种由ATP7B基因突变所致铜代谢异常的常染色体隐性遗传病,在世界范围内人群发病率为1/3万~1 /10万,致病基因携带者约1/90,我国该病的发病率比西方国家更高[4]。ATP7B基因位于染色体13q14.3上,编码一种P型铜转运ATP酶,它在肝细胞内起作用,使铜穿过细胞膜,产生铁氧化酶-血清铜蓝蛋白,并将铜排泄到胆汁中[5,6]。该病临床分型常分4型:肝型、脑型、其他型、混合型[4,7],其中绝大多数患者都是以肝脏和(或)神经系统受累为主要表现[8,9],其起病隐匿伴不典型临床表现常常增加疾病的诊治难度。
先证者首诊为亚急性起病,高度怀疑中毒代谢性疾病或感染性疾病。但相关检查未见异常。后结合患者年龄特点、磁共振示双侧基底节区异常信号,行HLD相关检查,最终确诊疾病。值得注意的是,HLD常表现为慢性起病,亚急性起病十分少见。据最近的一项研究显示,我国的364个肝豆状核变性患者中,约有32例是以亚急性起病的,构成比约为9%。有学者提出:起病形式可能与脑不同部位的损害有关[10]。有意思的是亚急性起病的患者的MRI常显示壳核和丘脑的损害,而慢性起病的患者不仅有上述部位的损害,还会有脑桥、中脑和皮质等部位的损害[10]。而损害常与星型胶质细胞和小胶质细胞的水肿、坏死有关,还可能与铜沉积所致的坏死和空洞有关[8,11],但损害部位的不同如何影响起病形式的具体机制仍待探讨。
在HLD患者中,由于铜沉积于神经系统,常导致锥体外系损害,临床上表现为肌张力障碍、共济失调等[11]。肌张力障碍是脑型HLD患者最常见的体征,以肌张力增高多见,而肌张力下降极其少见[6,9,12]。先证者表现为四肢肌张力下降,该种特殊体征的出现可能是游离铜沉积于脑组织基底节区域,聚集于星型胶质细胞的胞浆和溶酶体内,从而损害血脑屏障,导致轴突变性和神经细胞、少突胶质细胞变性坏死[8]。另一方面,游离铜可以直接损害功能蛋白,甚至改变相关基因的表达,或者产生ROS导致线粒体损伤并启动细胞凋亡[1,8,13]。至于神经源性细胞坏死或凋亡是如何引起不同的肌张力变化,仍然需要在未来继续探讨。
ATP7B 基因是目前唯一被广泛认可的肝豆状核变性的致病基因[4]。该基因编码P型铜转运ATP酶,参与铜的跨膜转运。该种酶由3类结构域组成:(1)铜离子结合域;(2)P型ATP酶结构域;(3)8个跨膜结构域[5]。研究显示,不同的基因突变常引起特定的临床表现[8,14]。例如,错义突变的患者临床表现较轻,进展较慢,而移码突变与之相反[15]。但若错义突变影响了重要的结构域,则症状严重[5,8]。先证者存在ATP7B基因的复合杂合突变(p.R778L;p.G943D),经检索pubmed数据库和基因库(http://www.wilsondisease.med.ualberta.ca/database.asp,http://www.hgmd.org/)等,证实此突变类型已被报道[16],为一名8岁儿童,但尚未有临床表现不典型的青年患者的报道。值得注意的是,先证者为ATP7B错义突变,影响了TM4和TM5跨膜区[17],这恰好是ATP7B蛋白的重要结构域,解释了疾病进展较快,结局严重(双下肢行走不能)的表现。有趣的是,研究提出:不同类型的基因突变可以很好地预测HLD的临床表型及发病时间[3,5,6]。但饮食、药物与环境等因素的作用不能忽略[18],故疾病预测模型的提出仍需进一步验证。
肝豆状核变性的临床表现错综复杂,加之首诊医生可能缺乏经验,从而导致误诊或延诊,耽误了早期治疗时间。因此,医务工作者需要重视HLD的不典型表现,在详细询问病史的基础上,行相关辅助检查,必要时进行基因检测,从而提高疾病的诊断和治疗效果。
