应用多重连接探针扩增技术检测X-性连锁鱼鳞病一家系STS基因
2021-07-22汤庄力牛新武耿松梅王晓鹏
党 阳 汤庄力 牛新武 耿松梅 刘 艳 王晓鹏
1西安交通大学第二附属医院皮肤病院,西安,710004;2浙江大学医学院附属第二医院,杭州,310009
X-性连锁鱼鳞病(X-linked ichthyosis, XLI, OMIM # 308100)由Lundborg等学者在1927年首次报道[1]。XLI患者几乎全部为男性,世界范围内的患病率约为每1500名男性中有一例患者[2],在所有型别鱼鳞病的构成比中排第2位。
XLI患者通常在出生后数周开始出现临床表现,但部分患者出生时即有鱼鳞病皮损[3,4]。皮损初发时表现为躯干四肢广泛分布的半透明大而细碎的鳞屑,随着病情进展,皮损逐渐发展为典型的深色多边形黏附性鳞片。皮损可累及四肢、躯干、颈部,褶皱部位亦可受累。此外,部分患者可同时伴有隐睾、角膜浑浊以及神经精神系统改变[5]。
XLI的致病基因为STS基因,该基因位于Xp22.31,共有10个外显子,基因长度约为135 bp,编码类固醇硫酸酯酶(steroid sulfatase, STS)。STS广泛分布于包括胎盘、皮肤、阴道、乳腺、血液、肝脏、脑等人体各组织器官[6],并被证实与XLI的发病机制相关[7]。本文报道1个呈现STS基因大片段缺失的X-性连锁鱼鳞病家系。
1 材料与方法
1.1 研究对象 先证者,男,31岁。出生时系足月顺产,生后生长发育同同龄儿。患者出生后2个月即出现全身皮肤干燥、粗糙,四肢伸侧为著,可见大小不一的多边形黑褐色鳞片。家系中无类似临床表现患者。先证者父母非近亲婚配。
1.2 外周血基因组DNA提取与质检 获取患者及其父母知情同意后,经肘正中静脉分别采集患者及其父母外周血2 mL,EDTA抗凝,使用全血基因组DNA提取试剂盒[天根生化科技(北京)有限公司]提取基因组DNA备用。
利用ND-1000-UV-VIS波长紫外/可见光扫描分光光度仪(Nanodrop,USA)对提取的基因组DNA进行定量检测。所提取的DNA样本浓度应介于20~100 ng/μL之间,OD260/OD280比值以及OD260/OD230比值均应介于1.8~2之间。
1.3 多重连接探针扩增技术检测 按照多重连接探针扩增P160-STS试剂盒(MRC,Holland)的操作说明书对所得基因组DNA样本进行多重连接探针扩增(multiplex ligation-dependent probe amplification,MLPA)。
将扩增所得的样本使用ABI3130型基因测序仪(Applied Biosystems,CA,USA)进行测序,运用MLPA分析软件Coffalyser对所得数据进行分析。
2 结果
患者所在家系中仅患者表现有XLI的临床症状,尤其是四肢伸侧可见大小不一多边形黑褐色鳞屑(图1)。
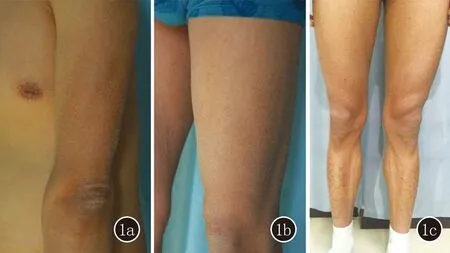
1a:左上肢伸侧皮损;1b:左下肢股骨前侧皮损;1c:患者双下肢皮损
对患者及其父母的MLPA分析结果可知,患者STS基因各外显子探针检测信号均为0,提示患者为STS基因缺失半合子。患者母亲STS基因各外显子探针检测信号均小于0.7,提示该基因全部外显子存在杂合缺失。患者及其母亲STS基因MLPA分析结果见图2。患者父亲STS基因各外显子探针检测信号均正常。
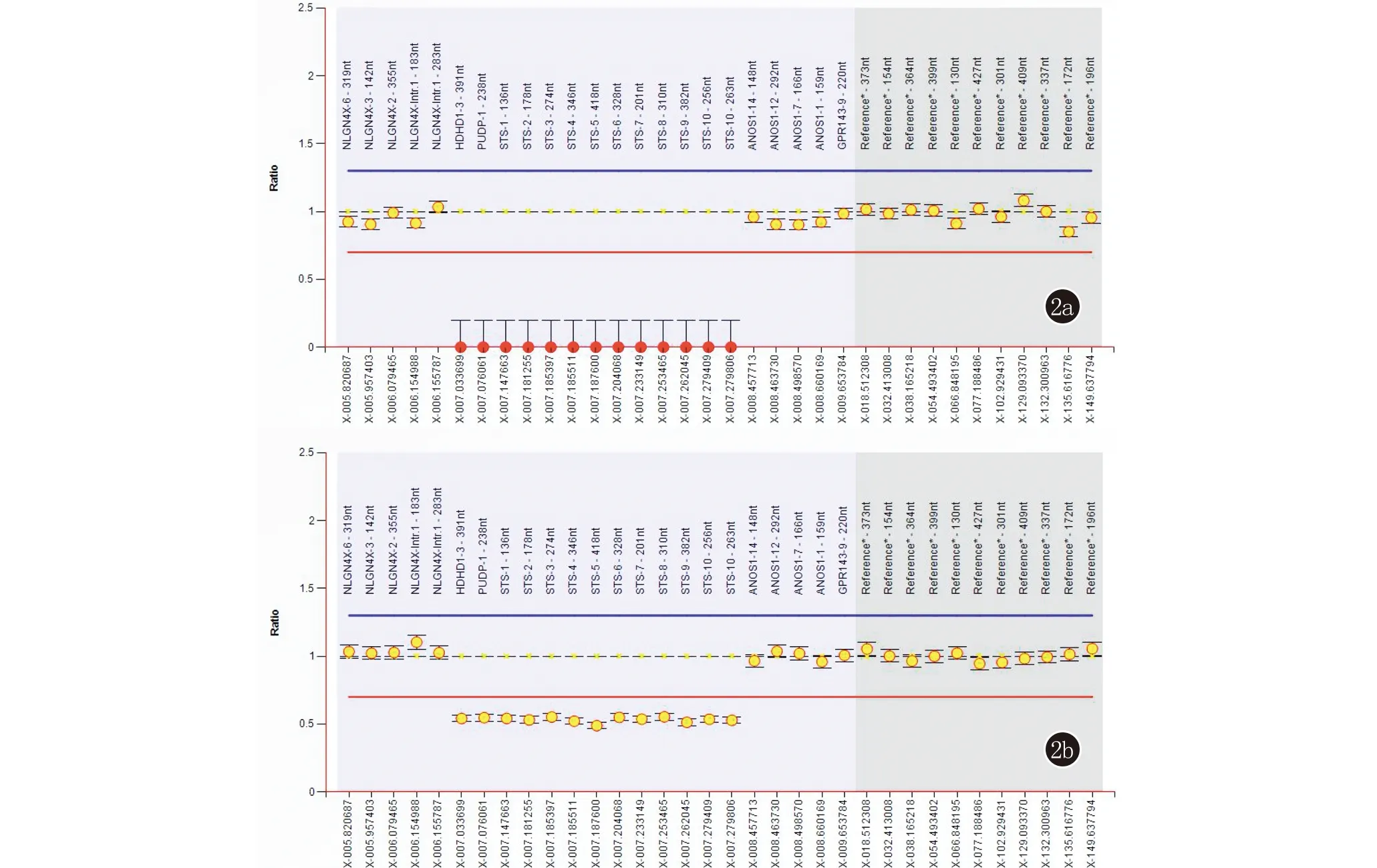
2a:患者STS基因MLPA结果图,该基因各外显子探针检测信号均为0,提示患者为缺失半合子;2b:患者母亲STS基因MLPA结果图,该基因各外显子的探针检测信号介于0.49~0.55之间,均小于0.7,提示患者母亲为缺失杂合子
3 讨论
XLI为伴X染色体隐性遗传性皮肤病。由于男性基因组核型为46,XY,一旦唯一的X染色体存在STS基因突变或缺失即可出现鱼鳞病的典型临床表现,而女性患者只有STS基因发生纯合突变或复合杂合突变时才会表现鱼鳞病的典型临床表现,截至目前,PubMed数据库中仅报道4例女性XLI患者[8],患病人数远远少于男性。
在健康人皮肤组织,STS主要存在于角质形成细胞的内质网膜上。当STS基因发生丧失功能突变时,其编码蛋白质结构及功能发生改变,局部胆固醇硫酸盐脱磺化过程受阻,一方面造成局部胆固醇硫酸盐的大量沉积,出现鳞屑等临床表现,另一方面造成胆固醇硫酸盐脱磺化的产物显著减少,影响皮肤正常的屏障功能,并改变皮肤正常的pH值。异常的皮肤pH值会影响局部丝氨酸蛋白酶的活性,表皮角质层细胞间黏附性增强,进一步出现表皮角化过度的病理生理改变[9]。
STS基因位于Xp22.31区域,属于人类X染色体上的进化层(evolutionary strata),该区域具有低丰度的散在重复序列(interspersed repeats)以及较低的GC含量,容易发生非等位基因同源重组[10,11]。该区域的基因除了STS外还有ANOS1、NLGN4X、HDHD1、PNPLA4和VCX。ANOS1基因突变可致卡尔曼综合征,NLGN4X和VCX基因与智力发育有关,而HDHD1和PNPLA4基因则分别与核苷酸去磷酸化以及维持表皮稳态相关[11]。本研究中的先证者在该区域发生了微缺失,造成STS以及HDHD1基因全部外显子缺失,但HDHD1基因突变所致人类疾病至今未见报道[12]。患者母亲STS基因各外显子的探针检测信号均小于0.7,但检测信号略有差异,值介于0.49~0.55之间,该基因各外显子检测信号的微小差异可能与莱昂现象(Lyon phenomenon)有关。
XLI为伴X染色体隐性遗传,该家系中患者的母亲无兄弟,姐妹拒绝接受相关检查,而患者的外祖父母已经过世,据其母回忆,患者的外祖父无鱼鳞病的相关临床表现,而STS基因发生原位缺失的几率小,因而推测该缺失杂合子可能来源于先证者外婆或更久远的亲代。
Xp22.31区域微缺失造成STS基因以及HDHD1基因外显子全部缺失在XLI较为常见,约占所有XLI患者的85%~90%[1],STS基因碱基替换或插入缺失突变仅占本病基因改变的很小一部分。运用MLPA技术常规开展产前筛查,尤其是在有鱼鳞病家族史的备孕夫妻中开展,对提高新生人口质量有较大帮助。
