拓扑半金属MoTe1.5S0.5与MoTe2电子结构和光学性质的比较研究
2021-07-16陈浩泽
王 胜,郑 鑫,陈浩泽,吴 林,成 立,张 莉
(中国计量大学 理学院,浙江 杭州 310018)
当前,能耗问题、量子隧穿与量子涨落效应等问题,从根本上阻碍了半导体电子器件的进一步微型化和集成化,成为现代信息和电子技术发展的瓶颈。探索和开发高效低能、突破量子尺寸效应的新一代量子材料迫在眉睫。拓扑材料由于具有独特的对环境细节(如缺陷、杂质甚至外界场、热、力等)不敏感特性,有望实现能量和信息的无损耗传播,产生基于新概念的电子和自旋器件、磁电和热电材料及器件、拓扑量子计算器件等,引起极大关注。2011年,南京大学的万贤纲与加利福尼亚大学的Savrasov、Vishwanath等人合作[1],通过理论计算首次提出在烧绿石结构的铱氧化物Re2Ir2O7(Re=稀土元素)中,可能实现外尔半金属态,并指出其特有的表面态——费米弧,开辟了拓扑半金属这一新研究领域。可见和普通金属相比,拓扑半金属的重要特征就是体能带在费米面附近有交点,且交点处的态密度为零。按照交点在动量空间的分布形态和简并度,拓扑半金属又可分为狄拉克半金属、外尔半金属和节线半金属等,如图1。
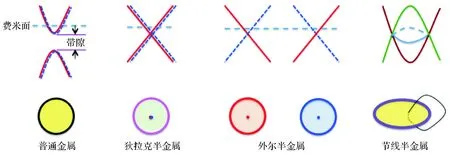
图1 拓扑半金属的分类[2]Figure 1 Classification of topological semimetals[2]
其中外尔半金属MoTe2凭借其丰富的物理性质(拓扑性,超导性)和优异的光电性能在过去十多年内引起了人们极大的研究兴趣[3-9],是拓扑超导体的有力候选材料。从晶体结构上看,MoTe2具有三种结构,分别为2H(六方),T’(单斜)和Td(正交)。在温度低于250 K时,1T’-MoTe2会转变为正交晶系的Td相,这个结构相变不改变面内的结构,而仅仅使层间的堆叠角由93.9°转为90°。与此同时,这种变化也使得Td-MoTe2破缺中心反演对称性[10-11],如图2所示(图中红色代表Te原子,绿色代表Mo原子)。较少的文献报道显示MoTe2掺S后其超导转变温度增加,例如文献[12]报道:MoTe2单晶的超导转变温度Tc为0.1 K,而MoTe1.8S0.2的Tc为1.3 K,可见掺S显著地提高了MoTe2的超导转变温度。但是,关于掺S后其光学性质的变化却鲜有报道。
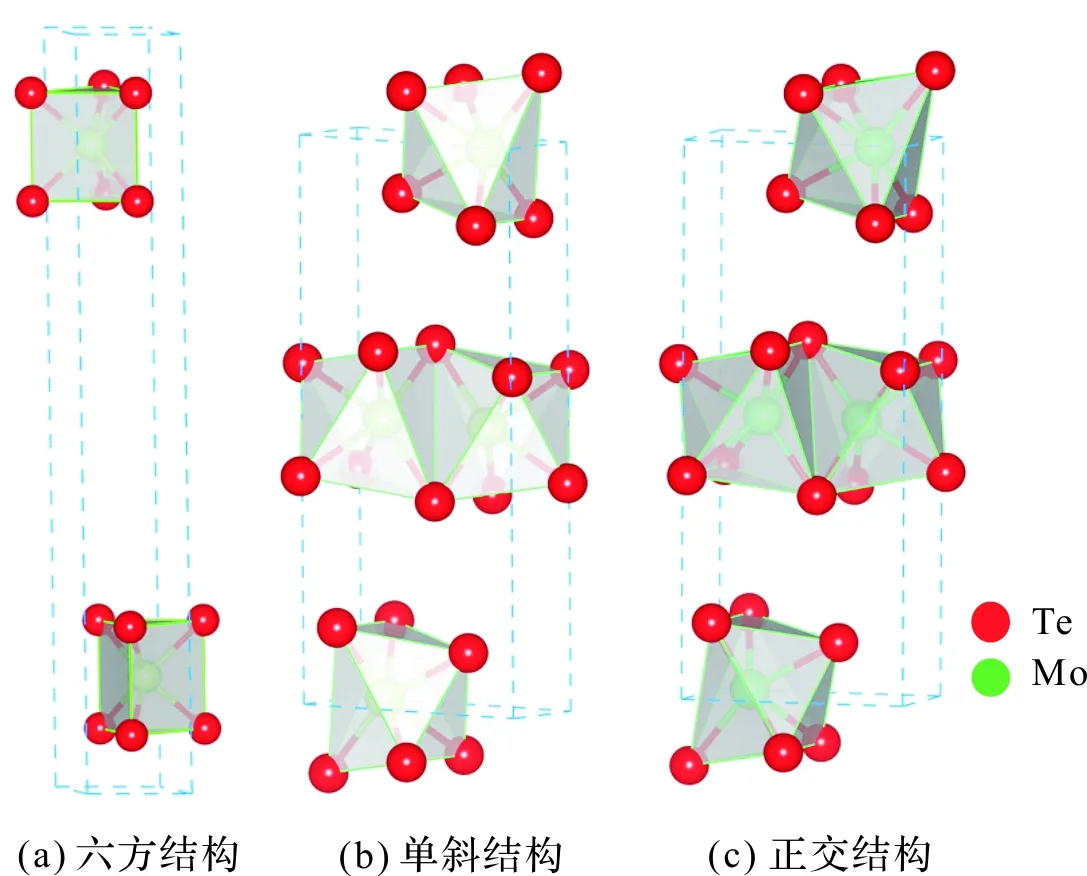
图2 MoTe2结构Figure 2 MoTe2 structure
本文根据第一性原理计算方法,利用MS软件的CASTEP模块对MoTe1.5S0.5与MoTe2分别建模,利用VASP软件包分别计算了掺S前后体系的电子结构和光学性质,研究了掺S后MoTe2电子结构对光学性质的影响,这些结果对从能带结构和光学性质角度理解拓扑半金属有一定参考价值。
1 模型构建和计算方法
采用Material Studio软件中的CASTEP模块建模,采用VSAP软件包计算能带结构和光学性质,采用计算效率较高的超软(ultra-soft)赝势考虑离子间的相互作用,采用GGA-PBE函数进行结构优化,分别计算了正交结构MoTe2(空间群为Pmn/21)及MoTe1.5S0.5的电子结构和光学性质。其中Mo、Te和S的价电子组态分别为,Mo[4s24p64d55s1]、Te[5s25p4]、S[3s23p4]。计算中优化参数的收敛标准依次为:单原子能量(Energy),1.0×10-5eV/atom;最大相互作用力(Max force),0.05 Å;最大内应力(Max.stress),0.1 Gpa;最大位移(Max.displacement),0.001 Å;截断能(Energy Cutoff),450 eV;Monkhorst-Pack特殊K点为12×10×6。通过查询ICSD晶体数据库,选取MoTe2优化前的晶格参数a=3.469 Å,b=6.36 Å,c=13.83 Å,Mo、Te的原子坐标见表1。
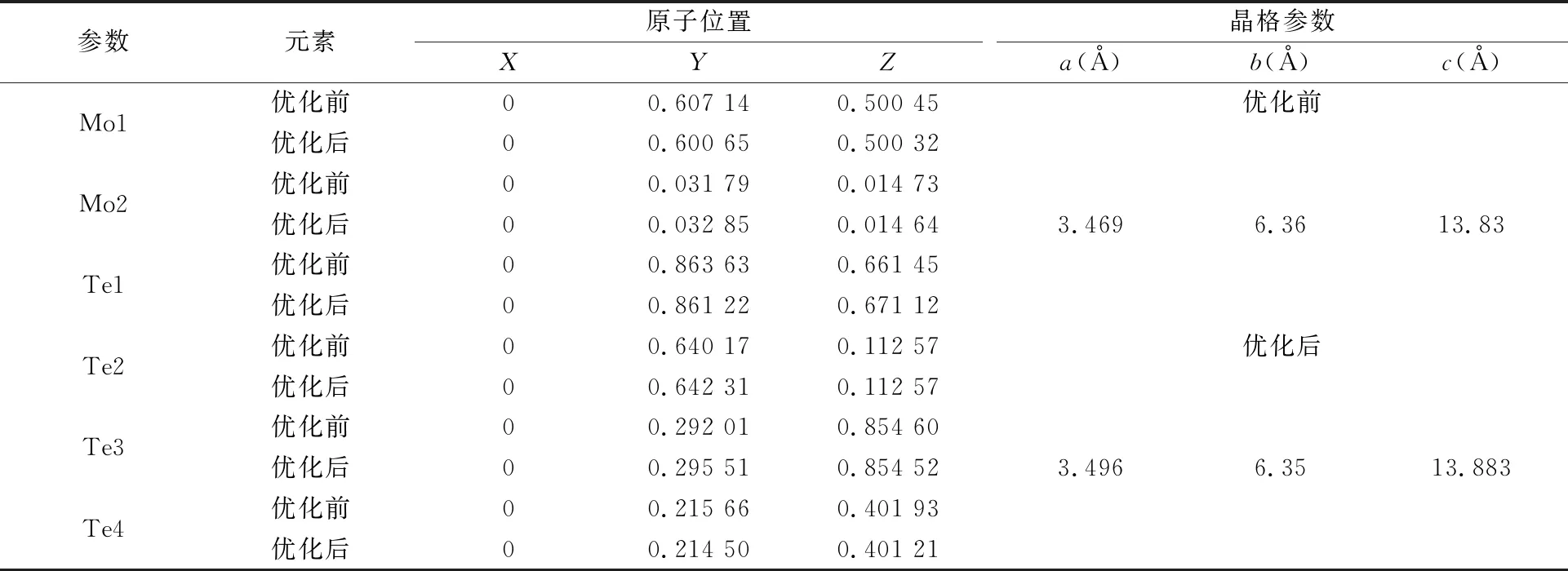
表1 优化前后各原子的位置以及晶格参数的变化
优化后MoTe2的晶格参数和原子坐标如表1,可见,优化后的晶格参数和实验值吻合的较好[13],表明选择的赝势和交换关联势可行。对于MoTe1.5S0.5体系,模型的构建如图3(b),S四种可能的掺杂位置分别用红、紫、蓝、黄表示,并分别计算了其对应的总结合能,结果表明:红色代表的掺S“位置一”能量最低,故后面关于MoTe1.5S0.5体系的电子结构和光学性质计算均采用“位置一”掺S的晶体结构。
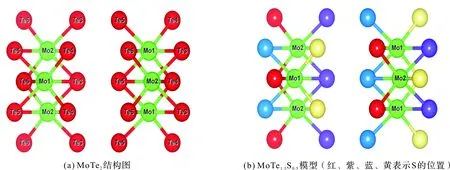
图3 MoTe2与MoTe1.5S0.5结构图Figure 3 Structural diagrams of MoTe2 and MoTe1.5S0.5
2 电子结构计算结果与讨论
图4(a)和(b)分别为计算所得的MoTe2和MoTe1.5S0.5的能带结构图,其中虚线为费米面Ef。可见,在高对称点G附近,无论是否掺杂,电子带和空穴带都穿过费米能级Ef,导带和价带之间的带隙较小,说明掺杂前后MoTe2的半金属性质并未改变。比较图4(a)和图4(b)显示:掺入S原子后,G点附近的能级结构下移,其价带顶从67 meV下移到-140 meV;带隙变小,从225 meV变成5 meV。仔细分析可能是S原子(原子半径为1.04 Å)代替Te原子(原子半径为1.70 Å)后,由于原子半径减小,从而引入化学压力所致。(1 Å=10-10m)
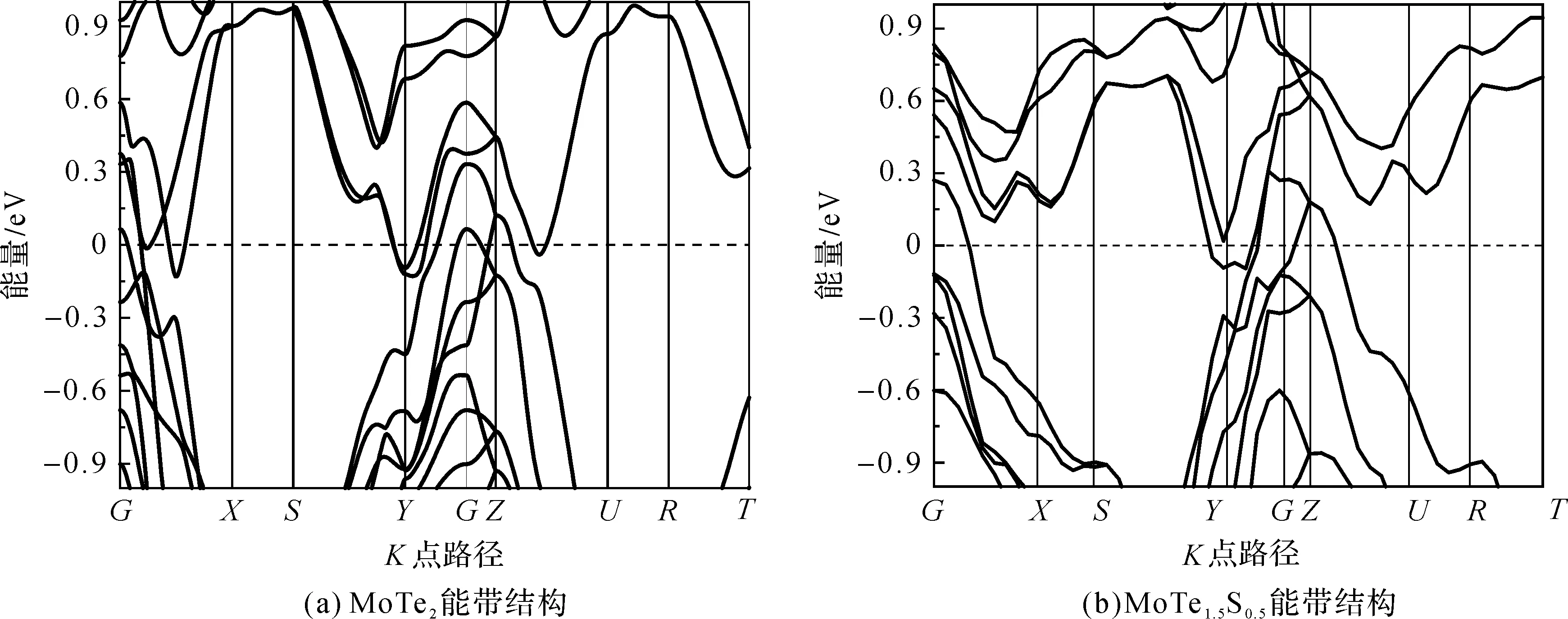
图4 MoTe2和MoTe1.5S0.5能带结构Figure 4 Energy band strustures of MoTe2 and MoTe1.5S0.5
由图5可知,费米能级附近,MoTe2的态密度主要由Mo的4d和Te的5p电子决定,掺杂后,S原子的3p电子对费米面有较小影响。仔细比较图5(a)和5(b),发现掺杂后,在总态密度图上位于费米面附近的最小态密度增加了0.1 eV,表明掺杂后形成的MoTe1.5S0.5结构稳定[14]。另外,图5(a)中,在-5.5~-3.5 eV,-3~-1 eV和1~3 eV能量区间,掺杂前Te的5p和Mo的4d轨道都出现了明显的峰值,这说明此区间Te和Mo形成p-d轨道杂化;图5(b)中,掺杂后Te的5p轨道能量在费米面附近略微减小0.1 eV,而Mo的4d轨道能量在费米面附近增加了0.5 eV,且在-6~-3.5 eV能量范围内,Mo的4d轨道和S的3p轨道都出现两个明显的峰值,这说明掺杂后S和Mo也可以p-d轨道杂化成键。
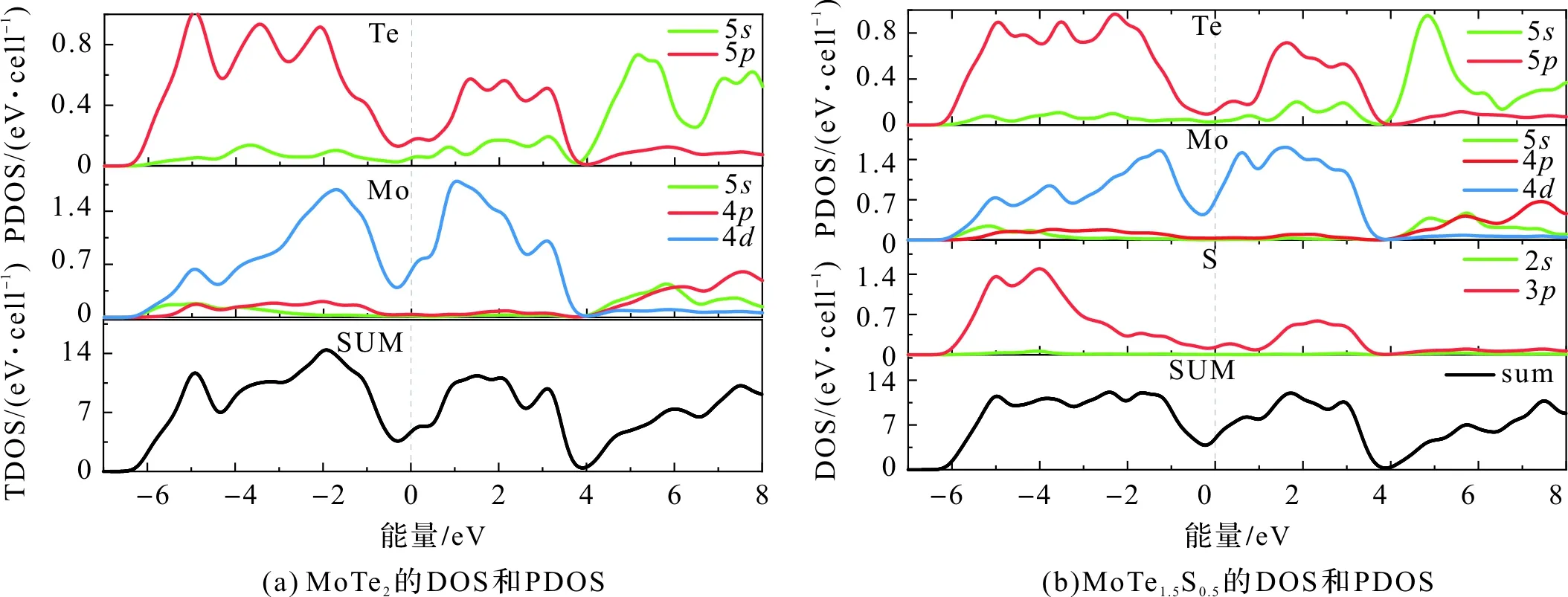
图5 MoTe2和MoTe1.5S0.5的态密度和分态密度Figure 5 Sum and partial DOS of MoTe2 and MoTe1.5S0.5
3 光学性质计算结果及讨论
介电函数描述系统对电磁辐射的响应效应,决定了晶体的主要光学性质。而其它光学性质如吸收系数、反射率等均可以根据直接跃迁概率理论以及Kramers-Kroning变换推导得出[15]。
我们首先计算了MoTe2单晶沿三个偏振方向([1 0 0][0 1 0][0 0 1])的光学性质(包括反射曲线、吸收曲线、光导率谱、复折射率曲线、介电函数和损失函数曲线),并分别画图比较之,见图6(a)至图6(f)。可见,MoTe2沿三个偏振方向([1 0 0][0 1 0][0 0 1])的光学性质总体差别不大。因此,后续对比研究MoTe2掺杂前后光学性质时,我们只选择[1 0 0]方向的计算结果进行比较。
总体来看,MoTe2掺S后沿[1 0 0]方向的反射率、吸收率、光导率的实部和虚部都略有下降,但掺S后损失函数峰明显增加、变窄和红移(见图7(a)至图7(f))。具体来看,图7(a)、图7(b)表明掺杂减少了反射率和吸收率,即增强了材料的透过率;图7(c)表明掺杂后折射率峰值向能量更高的区域略有偏移,但在可见光区域MoTe1.5S0.5的折射率高于母体的折射率;图7(d)表示损失曲线,掺杂后其峰值增加了33.33%,半宽度减小了16.67%,峰值位置略微红移(由21 eV变为20 eV)图7(e)表明,对于母体而言,光导率实部曲线存在三个峰值,分别位于1.52 eV、3.45 eV和7.80 eV;掺杂后,1.52 eV处的光导率峰消失,而位于7.80 eV附近的峰则一分为二;对于图7(f)而言,掺杂后在7.00 eV处出现一小峰。
特别地,由图7可见,对于反射率和复光导率的实部而言,无论掺杂与否,从零频部分变化都很剧烈。为了更好地理解掺杂对零频附近的影响,我们以波数为横坐标,在0~2.5 eV的范围内对反射率和复光导率的实部作图(如图8)进行比较。图8(a)中,母体在波数大于12 000 cm-1时,反射率变得较为平坦;但掺S后,反射率在波数小于2 500 cm-1时迅速下降,大于2 500 cm-1时则较为平坦;图8(b),掺杂后,光导率急剧变化后开始变缓的拐点也在2 500 cm-1附近。光学上把金属反射率急剧下降的区域称为“等离子边”,对应的频率称等离子频率,可见,MoTe1.5S0.5的等离子频率为2 500 cm-1。另外,对于纯MoTe2在5 000~7 000 cm-1光电导率几乎不发生改变,称之为“平坦的光电导率”。引入杂质后,这种“平坦的光电导率”向能量更高的方向移动,即出现在2 000~3 000 cm-1区间。
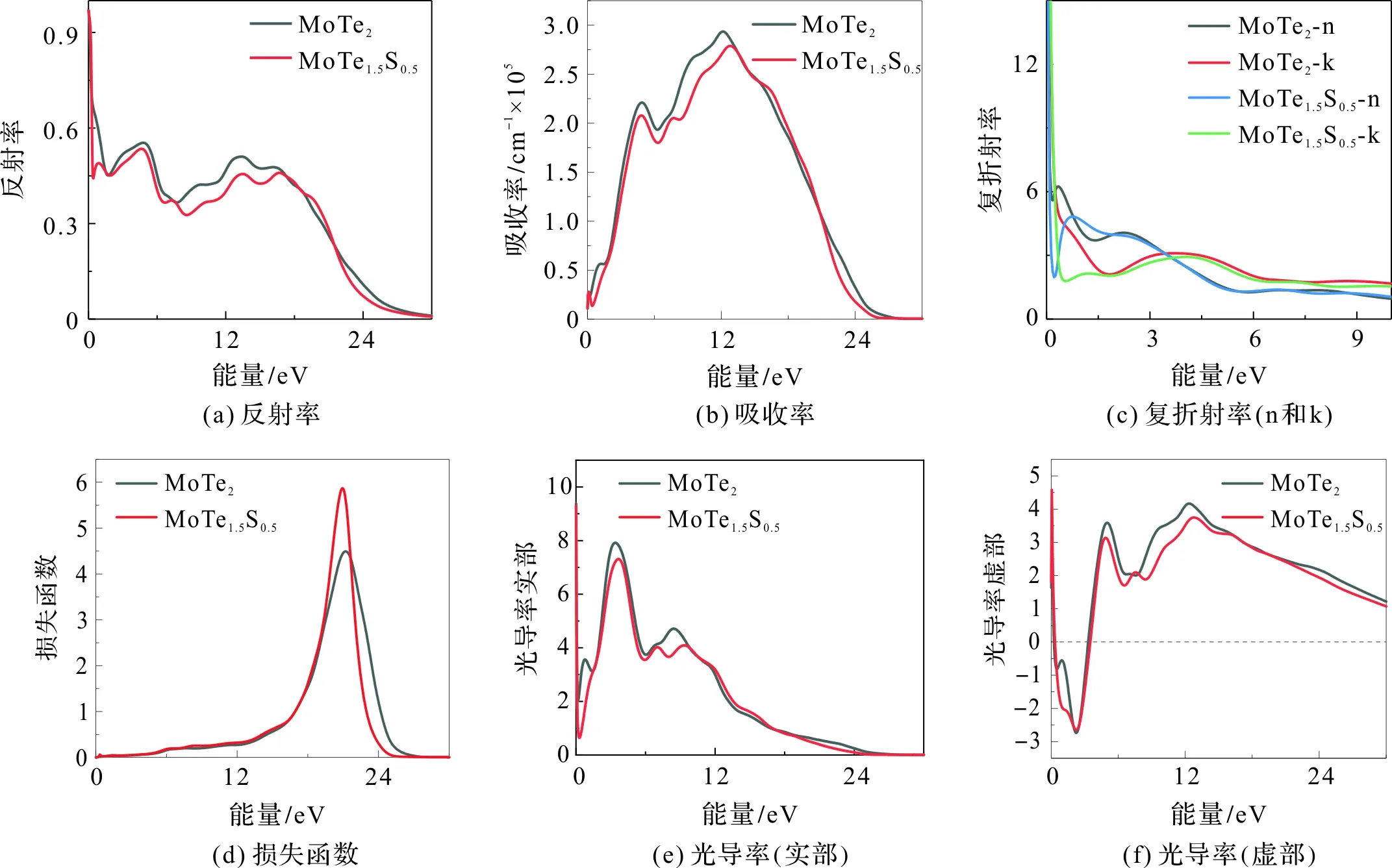
图7 MoTe2和MoTe1.5S0.5光学性质对比Figure 7 Comparison of optical properties of MoTe2 and MoTe1.5S0.5
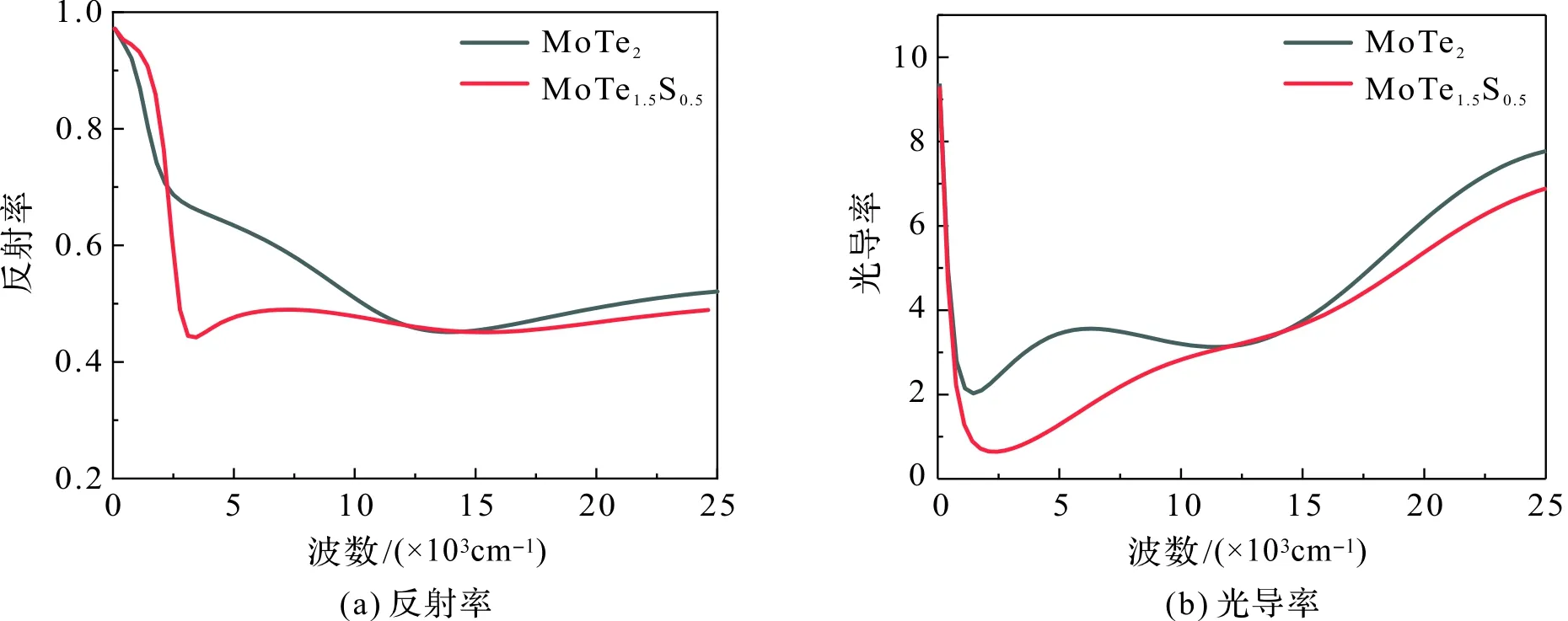
图8 MoTe2和MoTe1.5S0.5红外光谱区域对比Figure 8 Comparison of infrared spectral regions between MoTe2 and MoTe1.5S0.5
4 结 论
本文根据第一性原理计算方法、利用MS软件包的CASTEP模块对MoTe2和MoTe1.5S0.5分别建模,并验证了计算方法的可行性和掺杂位置的可靠性。然后利用VASP软件分别计算了掺S前后体系的电子结构和光学性质,仔细对比计算结果,发现1)掺S后电子结构变化:能带上G点附近的导带下移,带隙变小,分析得出这是由于S原子代替Te原子引入化学压力所致。(2)掺S后光学性质变化:MoTe2掺S后沿[1 0 0]方向的反射率、吸收率、光导率的实部和虚部均略有下降,但损失函数峰明显增加、变窄和红移。进一步分析反射率和光导率,发现MoTe1.5S0.5的等离子频率为2 500 cm-1,对于纯的MoTe2在5 000~7 000 cm-1光电导率几乎不发生改变,称为“平坦的光导率”。引入S杂质后,这种平坦的光电导率,向着能量更高的方向移动,即出现在2 000~3 000 cm-1。这些结果对从电子结构和光学性质角度理解拓扑半金属有一定参考价值。
