欧盟生产质量管理规范监管制度对我国药品生产企业的启示*
2021-07-15靳玉瑶赵利斌
靳玉瑶,赵利斌△,王 娟
(1. 北京振东光明药物研究院有限公司,北京 100085; 2. 山西中医药大学研究生院,山西 晋中 030619)
2011 年,我国新版药品《生产质量管理规范》(GMP)实施以来,执行标准与欧盟GMP 不断接轨,我国制药企业生产质量也随之不断提升。近年来,我国通过欧盟GMP 认证的企业数量也越来越多,但多为原料药生产企业,制剂生产企业较少。可见,我国制剂生产企业还应不断与国际标准接轨,加速国际化进程。为此,综述了欧盟GMP 体系的现状,分析我国药品生产企业(简称药企)欧盟GMP 认证的现状,并提出了改进措施。
1 欧盟GMP 体系
1.1 监管体系
欧盟作为欧洲国家的政治经济联合体,对医药产品采取了统一的管理措施,所有成员国均具有相同的法规及GMP。欧盟要求所有医药产品的生产及进口必须通过许可,授权持有人必须遵守GMP 的原则和要求,并使用符合欧盟GMP 的车间生产药品。对于GMP 检查,欧盟各国有相同的质量控制要求,且欧盟内部时常组织多个国家联合审计,确保检查监管水平的一致性。欧洲药品管理局(EMA)与欧洲药品质量理事会(EDQM)是欧盟2 个不同的药品监督管理机构。EMA 受理制剂产品的注册和GMP 监管。EDQM 是欧洲药典(EP)及标准品的发布机构,并负责原料药的欧洲药典适用性证书(CEP)申请注册及 GMP 检查。
欧盟为了解决欧洲自由贸易联盟国家之间的药品贸易问题,并促进会员国之间的药品贸易,于1970 年加入国际药品认证合作组织(PICS)。截至2017 年3 月,美国食品药物管理局(FDA)完成了对28 个欧盟成员国药品检查机构能力的评估,与欧盟已达成互认协议。自2017 年11 月1 日起,欧盟成员国与FDA 均不会重复检查对方已执行的检查,包括已上市的各种人用药物制剂(如片剂、胶囊剂、软膏剂、注射剂等)、已上市的生物制品、中间体、活性药物成分及原料药、兽药。预计到2022 年,人用疫苗、血浆衍生物也将被纳入其中。
欧盟对GMP 的管理非常严谨,1972 年颁布了《药品生产质量管理规范总则》,用于指导欧洲共同体(简称欧共体)国家的药品生产,1983 年进行了大幅修改。1989 年,第1 版欧盟GMP 出版,收载于欧盟药物管理规则和规章 EudraLex - Volume 4[1],并不断进行补充和修订。详见表1。

表1 1965 年至2017 年欧盟GMP 相关法规准则Tab.1 List of EU GMP relevant regulations and guidelines from 1965 to 2017
欧盟GMP 的现场检查由欧盟各成员国的专职检查员承担[2-3],制剂制造商或进口商必须获得授权才能制造或进口产品,且必须经过GMP 检查,检查频率一般为2 ~3 年 1 次。原料药的 GMP 检查基于风险评估,无风险或风险非常小的原料药无需进行GMP 检查,若产品有风险(如无菌原料药),或历史上检查有重大缺陷,或产品有过投诉、召回、大量退货等,会被重点检查。
1.2 独特的受权人制度
欧盟自1975 年开始实施受权人制度,这种制度能有效保证企业各级人员履行质量职责,保障药品质量,现已成为欧盟GMP 体系的核心。受权人是独立于生产、质量管理体系之外的第三方监控体系,主要对企业的质量管理体系进行审计及监控。受权人由欧盟指令及成员国法律授权,对药品质量承担最终责任,法律地位非常高,可由独立于生产部门、质量控制部门以外的企业全职人员或外聘专职受权人担任。欧盟2001/83/EC 指令对受权人管理作出了全面、原则性要求[4]:第41 条规定,药品生产许可的申请人至少必须有1 名受权人为其服务;第48 条规定,各成员国应采取各种有效措施,以确保生产许可持有人长期、持续地拥有至少1 名受权人的服务;第51 条明确规定,受权人要对每批药品生产的合法性及产品质量负责。受权人需要负责审核药品生产相关的所有重要质量标准、工艺规程、验证方案和报告、检验方法等,保证生产的产品符合欧盟标准,并与申报资料保持一致,且会定期对药厂进行检查。在GMP 指南附录16[1]中,也对受权人确认及批放行作了详细规定:受权人要对每批产品生产的合法性、合规性进行确认,并对生产管理过程、质量管理过程及该批产品质量有关的其他一切因素进行监管[5]。对于不能完成职责的受权人,成员国可通过行政管理或惩戒手段对该名受权人进行暂时停职处理[3]。
2 我国与欧盟GMP 制度的差异
欧盟注重动静结合的控制,不仅对车间体系等静态管理要求严格,对生产过程的每个步骤都严格要求,我国则更注重静态控制;欧盟对无菌产品的要求较高,所采用的一些专业技术,如隔离操作和吹灌封技术我国还未涉及;欧盟在GMP 认证时通常分为体系认证和产品认证,我国强制进行GMP 体系认证,但药品认证方面还有所欠缺;欧盟审计工作更注重防患于未然,要求对所有可能发生的隐患要做出预案,且对质量风险管理具有前瞻性和回顾性,我国还不够完善,仍需改善[6-9]。
3 我国与印度药企的欧盟GMP 认证现状分析
3.1 接受欧盟GMP 检查情况比较
截至2019 年 12 月 31 日,我国药企共接受欧盟GMP 检查516 次(按检查结束日查询),获得欧盟GMP认证合格证书473 个,不合格声明43 个,不合格率为8.33%。2010 年后,我国药企获得欧盟GMP 证书的数量持续增加,2018 年接受检查次数最多(92 次),表明欧洲市场对中国生产原料药及制剂产品的需求在不断增加;2016 年起,我国药监部门开始实施“双随机”突击检查及不告知飞行检查政策,且力度不断加大,国内药企GMP 管理体系的完善程度和实施水平明显改善,通过国际高端GMP 认证的能力显著提高,我国收到欧盟GMP 检查不合格声明的比例也大幅下降。印度药企接受欧盟GMP 检查共783 次,获得GMP 认证合格证书744 个,不合格声明 39 个,不合格率为 4.98% 。2005 年至2019 年接受检查的次数逐年上升。详见表2。可见,印度接受检查的次数及获得GMP 证书的数量均远超我国,且不合格率较我国低。我国接受欧盟GMP 检查的药企以原料药生产企业为主,制剂生产企业数量在近几年有所增加。印度接受检查的制剂生产企业总数已超过原料药生产企业,尤其是从2017 年开始,接受欧盟GMP检查的制剂生产企业数量迅速增加。中印两国均为人口大国,也是药品出口大国,我国应加快制剂生产企业国际认证的速度,为我国制剂产品的出口提供保障。
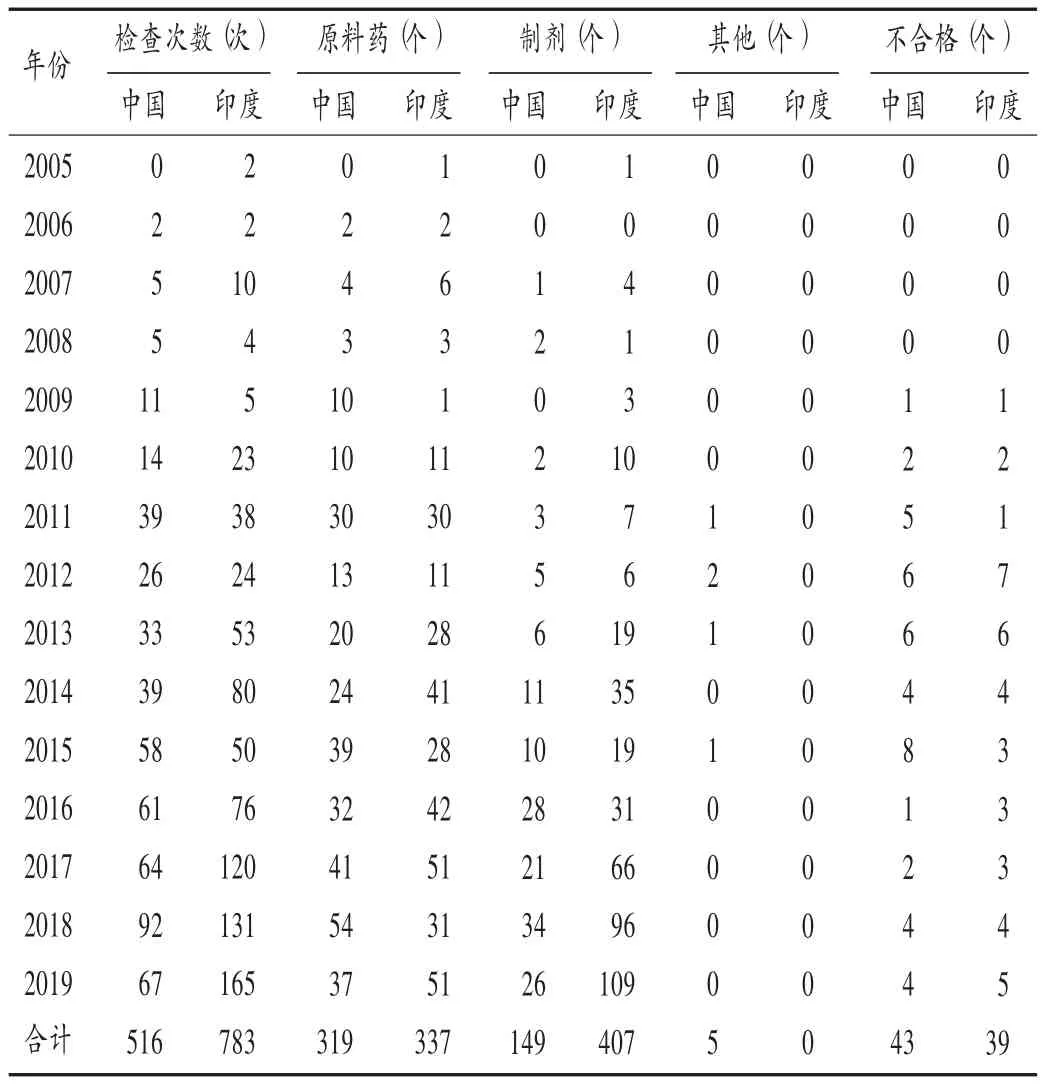
表2 2005 年至2019 年中国和印度药企接受欧盟GMP检查情况Tab.2 Statistics of Chinese and Indian pharmaceutical manufactures accepting the EU GMP inspection from 2005 to 2019
3.2 主要缺陷分析
EudraLex- Volume 4 规定了 EC 指令 2003 /94 /EC和91/412/UNECE 中规定用于人类和兽用药品生产管理规范的原则和底线。各成员国根据此卷规定对药企进行检查,对不符合欧盟GMP 要求的药企发布不合格声明[10]。截至 2019 年 12 月 31 日,我国共收到 43 个不合格声明报告,去除1 个重复报告,共有42 份有效报告。其中包括原料药生产企业39 个,制剂生产企业1 个,其他企业2 个。经分析发现,企业存在1 项重大缺陷,或者4 项以上主要缺陷,则会被认定为不符合欧盟GMP 标准。
原料药生产企业:共收到不合格声明39 份,占原料药生产企业总数的11.93%,其中24 份声明中包括重大缺陷,占声明总数的61.54%。重大缺陷最多的方面为质量管理,包括质量保证系统缺陷、质量控制系统缺陷、供应链质保监管不佳、储存环境控制不当、药品稳定性不佳等;其次为污染方面,包括交叉污染、清洁不当导致的杂质污染、微生物污染、对污染的管理措施不足等;记录方面,包括重要数据缺失、数据伪造、数据无追溯性、无偏差记录等;设施设备方面,包括不足以支持药品生产、损坏、设计缺陷、维护与清洁不当。主要缺陷涉及广泛,除重大缺陷中包括的质量管理缺陷、污染缺陷、记录缺陷、设施设备缺陷外,还包括仪器校正、标准品管理、工艺验证、偏差管理、供应商资质、变更控制、对投诉的管理、纯化水系统、物料管理、关键人员资质、标签标识不准确或缺失、电子原始数据保存不当、混批不规范等方面。可见,质量管理是原料药生产中最重要的部分,也是最容易出现重大缺陷的部分;其次为生产记录,应保证数据的真实性、完整性和可追溯性,禁止数据造假;此外,厂房设施设备、上游供应商资质、工艺等也为重要检查部分。
制剂生产企业:共得到不合格声明1 份,占总制剂生产企业的0.67%。重大缺陷体现在产品货架期质量不稳定,出现变色现象,且未对质量不稳定原因进行调查;此外,也未进行市场剩余产品的风险评估。因此,在产品生产中,不仅要重视车间的体系建设,还应重视产品的质量管理。
4 对我国药企的启示
4.1 重视车间硬件设施建设
欧盟认为,高标准的硬件可避免人员操作的随意性[11]。车间建设时需选择国内领先技术水平、满足欧盟标准的设备,且需通过相关技术人员对设备型号、材质等严格把关。车间应严格按欧盟GMP 要求进行建设与改造,如空调系统、空压系统、水系统等,不仅要做好工艺布局的审核,还要做好施工质量的检查验收,并做好相关记录[12]。
4.2 重视文件记录
欧盟官方对药企进行GMP 现场检查的时间仅有4 d,但需要检查整个GMP 体系文件。故企业要满足GMP 要求,需要进行硬件与软件改造,充分做好前期准备工作,完善质量管理体系的建设及文件整理。检查中最重要的一部分即为文件记录,记录缺失、伪造等都会给企业的GMP 体系建设、信誉度及其他药品的国内外销售造成严重不利影响,且短时间内不易消除[13]。因此,企业应高度重视文件记录的完整性、真实性、及时性和可追溯性。
4.3 重视产品风险评估
欧盟GMP 检查注重防患于未然与风险评估。只要数据与文件规定及生产条件不符,都一律视为偏差,所有偏差都应调查清楚,并启动纠正与预防措施程序,在确认对产品质量没有影响后才可放行[14]。质量保证人员应对偏差情况进行汇总和分析,并将其作为培训的主要内容,从中吸取教训,积累经验,避免相同偏差再次发生。
4.4 重视生产全过程质量控制
欧盟质量监管重视药品溯源性,药品生产过程中使用到的原料、辅料、药品包装材料等,下游企业均需追溯上游企业,确保药品的最终质量,尤其是我国的中药产品,成分复杂,作用机制不明确,活性物质不确定。欧盟规定,符合要求的传统草药进行欧盟注册时可适当减免临床及非临床试验,但药品质量与传统使用历史无关,需要从药材开始,到生命周期结束,全生产链控制药品质量[15]。
4.5 加强制剂生产企业国际认证
目前,我国通过欧盟GMP 认证的制剂生产企业数量仍较少,远不及我国原料药生产企业和印度制剂生产企业。为使我国制剂产品快速走出国门,应加大力度进行制剂生产企业的国际注册[16]。制剂生产较原料药周期长、过程复杂,应更加重视生产过程的质量管理。另外,还应加强产品货架期管理,保证药品整个生命周期的质量稳定性。
