富B酸氧化铝的制备及以其为载体催化剂的加氢脱硫反应性能
2021-07-14赵振祥张文成吴萍萍刘晨光阎子峰
赵振祥,刘 宾,王 丹,张文成,吴萍萍,刘晨光,白 鹏*,阎子峰
(1.中国石油大学(华东)化学工程学院重质油国家重点实验室,山东 青岛 266580;2.中国石油石油化工研究院大庆化工研究中心,黑龙江 大庆 163714)
随着油品消耗量的增加,环境污染也日趋严重。近年来,各国对油品中硫含量进行了严格限制。2009年,欧盟规定车用柴油的硫含量必须满足欧Ⅴ排放标准,即硫质量分数在10×10-6以下。我国在2020年实施了国六排放标准。尽管目前炼油企业基于现有催化剂体系已经生产出满足国六标准的柴油产品,但柴油脱硫深度的提高,造成加氢反应苛刻度提高,最终导致能耗、氢耗和生产成本升高。同时苛刻的反应条件也造成催化剂稳定性下降,运行周期变短。因此,开发高性能的柴油超深度脱硫催化剂是发展高效、低成本清洁柴油生产技术的关键。
柴油超深度脱硫的难点在于多取代基强空间位阻的多环硫化物,如4,6-二甲基二苯并噻吩(4,6-DMDBT)中硫的脱除如图1所示。
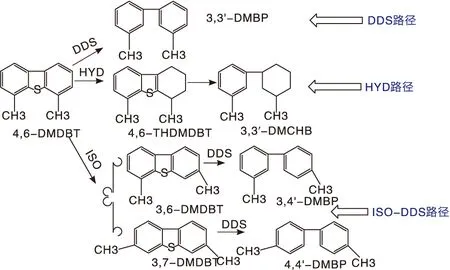
图1 具有强烷基位阻硫化物(4,6-DMDBT)的脱除路径图Figure 1 Hydrodesulfurization path of sulfide with strong alkyl steric hindrance (4,6-DMDBT)
该类硫化物的脱除存在三条可能的路径:(1)加氢脱硫路径(HYD)。HYD路径是先将其中一个苯环进行加氢饱和后,再将S原子进行脱除。通过HYD路径实现4,6-DMDBT有效脱硫,需要提高加氢脱硫反应的苛刻度,如提高氢分压,降低空速,提高反应温度,但造成设备投资加大,氢耗上升,操作成本提高,催化剂稳定性下降。(2)直接脱硫路径(DDS)。由于强烷基位阻的影响,4,6-DMDBT中硫原子以DDS反应路径脱除非常困难。Wang H等研究发现NiMo/γ-Al2O3上4,6-DMDBT的加氢脱硫主要以HYD路径为主,且HYD路线的反应速率是DDS路径的12.5倍[1],但DDS路径氢耗少。通过优化催化剂,增加DDS反应速率将降低超深度加氢脱硫反应成本,并优化产物分布;因此,研究者提出了第三条路径。(3)异构-再直接脱硫路径(ISO-DDS)。即先实现4,6-DMDBT中的烷基转移,降低硫原子的空间位阻,使其更易经DDS路径脱除。相比之下,三条反应路径中,ISO-DDS路径具有反应苛刻度低,4,6-DMDBT脱除效率高等优势。因此,经济而有效地实现柴油超深度脱硫的关键,在于设计新型的高效加氢脱硫催化剂,使4,6-DMDBT选择性地按照ISO-DDS路径进行脱除。
ISO-DDS路径强化的关键是高效催化材料,尤其是新型载体材料的设计开发。为了让这类化合物中具有强位阻的硫原子更易接近催化剂的活性位,载体应该具有一定的异构化活性,使这类分子中硫原子可接近性增加,比如将4,6-DMDBT异构成3,6-或者3,7-DMDBT(见图1)。Michaud P等研究发现,B酸能促进大分子硫化物的烷基转移,将其转化成空间位阻较小的异构体,使硫原子更易接近催化剂活性中心,进而提高其脱除效率[2]。为此研究者们尝试将含有B酸的分子筛加入催化剂中。结果表明,B酸促进了甲基转移反应,生成了空间位阻较小的3,6-或者3,7-DMDBT,大大提高了ISO-DDS路径的反应速率[2-4]。但分子筛一般具有较强的酸性位,在反应过程中不可避免地促进裂化以及聚合等副反应,造成柴油收率下降和气体产物增加[2,5-7]。Kwak C等研究表明,氟原子可以调变氧化铝的表面酸性,提高催化剂HDS活性,尤其是强化了对空间位阻较大的硫化物(如4,6-DMDBT)的脱除[8]。Kim H等研究表明,采用NH4F改性氧化铝载体后,4,6-DMDBT的HYD和ISO-DDS脱除路径的反应速率都有所增强。吡啶吸附红外的结果也表明NH4F改性能在载体表面产生B酸[9],但该方法不能有效调控氧化铝表面的B/L酸值。
本课题组前期研究结果表明[10-13],氟硼酸铵能有效地调节氧化铝的表面酸性,其不仅可以降低L酸还可增加B酸,实现表面酸性的大范围调变,并得到富含B酸的氧化铝材料,这为系统性调控B/L值提供了一种思路。本文以拟薄水铝石为铝源,以氟硼酸铵为改性剂,合成具有不同B/L值的改性氧化铝材料,并以改性氧化铝材料为载体制备加氢脱硫催化剂,评价探究B/L值对4,6-DMDBT超深度加氢脱硫路径选择性的影响。
1 实验部分
1.1 实验原料
氟硼酸铵(分析纯),上海三爱思试剂有限公司;拟薄水铝石,山东烟台恒辉化工有限公司;钼酸铵(分析纯),国药集团化学试剂有限公司;硫化铵溶液(40%~48%),上海阿拉丁生化科技股份有限公司;硝酸钴(分析纯),上海麦克林生化科技有限公司;4,6-二甲基二苯并噻吩(分析纯),上海麦克林生化科技有限公司;甲苯(分析纯),国药集团化学试剂有限公司。四硫代钼酸铵为自制,以钼酸铵为原料,在氨水中溶解后加入足量硫化铵溶液,80 ℃反应1 h,过滤并用蒸馏水洗涤,最后使用乙醇洗涤。
1.2 样品制备
将拟薄水铝石在550 ℃下焙烧4 h后,在氟硼酸铵溶液中40 ℃下浸渍2 h,干燥后得到未焙烧的氟掺杂氧化铝,命名为F/Al-x(x为氟铝物质的量比),500 ℃下焙烧4 h得到焙烧后的氟掺杂氧化铝,命名为CF/Al-x(x为氟铝物质的量比)。
为了消除活性相的影响,采用文献报道的制备方法使催化剂具有近似相同的活性相结构[14]。改性样品浸渍四硫代钼酸铵和硝酸钴的溶液后,经500 ℃焙烧3 h得到氧化态催化剂,氧化态催化剂经硫化铵溶液浸渍还原后在氮气氛围中100 ℃烘干3 h得到还原态催化剂。
1.3 样品表征
采用德国布鲁克公司D8 Advance X射线衍射仪对样品进行物相分析。工作电流 40 mA,扫描范围5o~75o,Cu Kα,入射射线波长0.1542 nm。
采用安东帕-康塔公司Quadeasorb evoTM全自动比表面积和孔隙率物理吸附仪对氧化铝的孔结构进行分析。测试前所有样品在300 ℃下进行真空脱气处理,然后以高纯氮气为吸附质,以液氮为冷阱在-196 ℃条件下测定样品的吸脱附等温线。比表面积采用BET法计算,孔径分布以BJH法计算,孔容以最大相对压力对应的吸附量进行计算。
采用扫描电子显微镜上的能量散射X射线荧光光谱(EDX,型号EDX-720并与日本电子JEOL生产的JSM-7900F热场发射扫描电子显微镜组合使用)对样品中的元素含量进行分析,测试电压10 kV,每组样品选取三个区域对表面F和Al元素含量取平均值。
采用德国布鲁克公司VERTEX 70V红外光谱仪对样品进行吡啶吸附透射红外检测。将样品于300 ℃干燥3 h除杂,采集背景后,置于装有吡啶的原位池中真空吸附,之后在150 ℃脱除物理吸附的吡啶;按公式1计算L酸量,以公式2计算B酸量。
(1)
(2)
Lacid为L酸量;SL为1 450 cm-1处的峰面积;R为样品槽的直径;W为样品的平均质量;Bacid为B酸量;SB为1 540 cm-1处的峰面积。
1.4 4,6-DMDBT加氢脱硫反应评价
通过降低氢气分压在转化率低于10%的前提下,探究氧化铝表面酸性对脱硫路径的影响。使用100 mL的高压反应釜作为反应器,以硫含量1%的4,6-DMDBT-甲苯溶液作为进料,进料量40 mL,催化剂用量0.5 g,反应温度260 ℃,氢气压力1 MPa,反应时间10 h。
2 结果与讨论
2.1 材料表征结果
图2为改性氧化铝的吡啶吸附红外光谱图,其表面酸性数据见表1。
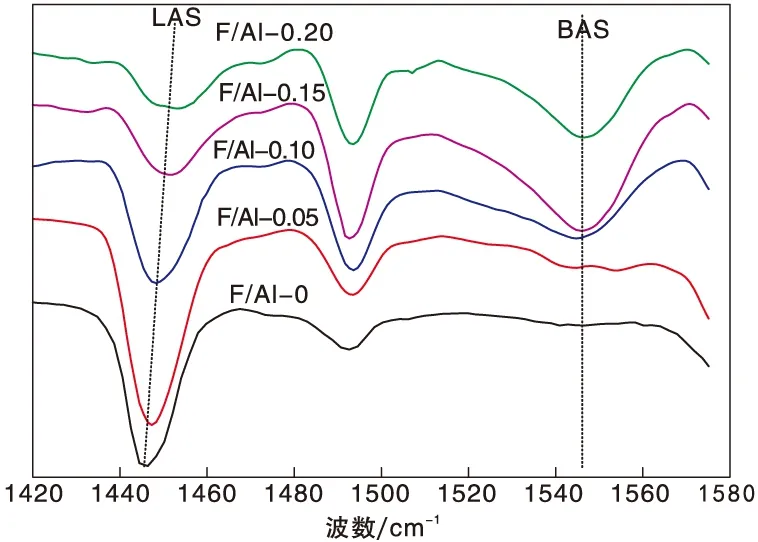
图2 改性氧化铝的吡啶吸附红外光谱图Figure 2 FT-IR spectra of pyridine adsorption on alumina samples
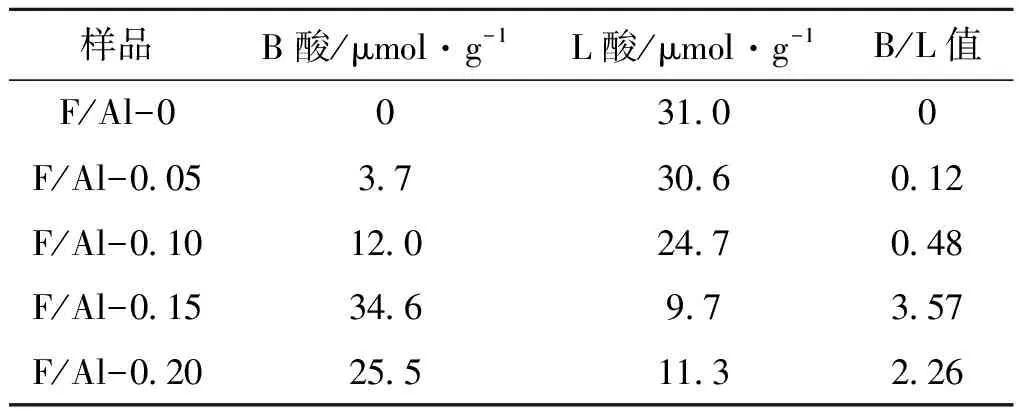
表1 改性氧化铝的表面酸性数据
从图2可以看出,1 545 cm-1附近的振动峰为吡啶吸附于B酸上引起的特征峰,1 445 cm-1附近的振动峰为L酸的特征峰[15-16]。与文献报道结果一致,氟的引入使氧化铝表面形成了B酸[17-18],并且随着掺杂量的增加,在1 545 cm-1处与B酸有关的特征峰明显增强。由表1可知,随着掺杂量的增加B酸含量先增加后减少,在F与Al物质的量比为0.15时B酸量达到最大值,且此时L酸量最少,B/L值最高为3.57。课题组前期研究结果表明[11],过高的氟含量会使氧化铝趋向于形成AlxFy[19],使得L酸量不减反增。与前期工作相比,在本工作中,F与Al比例降低了4倍,而B/L值提高了3.5倍,这是由于先焙烧后浸渍提高了氟硼酸铵的改性效率。
在之前的研究中[11],课题组采用了溶胶-凝胶法,向胶溶后的拟薄水铝石中加入改性剂,经老化后改性剂既存在于氧化铝表面也存在于体相中,改性剂进入体相可能是影响改性效果的主要原因。为了探究氧化铝表面氟铝比对改性效果的影响,对焙烧前后的改性氧化铝做EDX分析,结果如表2所示。

表2 焙烧前后氧化铝表面F/Al值的变化Table 2 Change of F/Al ratio on alumina surface before and after calcination
由表2可知,焙烧前样品中F/Al(物质的量比)的实际测量值是理论值的2倍以上,这说明氟物种主要存在于改性氧化铝的表面,有利于提高氟的改性效率。但焙烧后,F/Al(物质的量比)会降低。当F/Al(物质的量比)从0.05提高到0.15时,F/Al(物质的量比)实际值与理论值的差距由2倍降低到1.6倍,这说明一方面焙烧操作会引起氟的挥发流失,另一方面氟会进入氧化铝的体相结构中,造成氟的利用率降低。在我们前期的研究中也发现未经洗涤的样品改性效果更好,这主要是因为洗涤造成氟的流失[12]。与此相似,焙烧可能会使部分氟离子流失,并使构建的B酸消失。
为了进一步证明这一推断,将改性后的氧化铝经550 ℃焙烧后测试吡啶吸附红外,结果如图3所示,计算得到的L酸和B酸量数据见表3。由表3可以看出,随着掺杂量的增加,L酸量逐渐降低,B酸量先增加后降低,当F/Al(物质的量比)为0.15时B/L值具有最大值,这与焙烧前样品具有相似的变化规律。但与焙烧前样品相比,焙烧后氧化铝的L酸和B酸均降低,根据元素分析结果推断这主要是因为焙烧会影响氟在氧化铝表面和体相中的比例,从而影响改性效果。
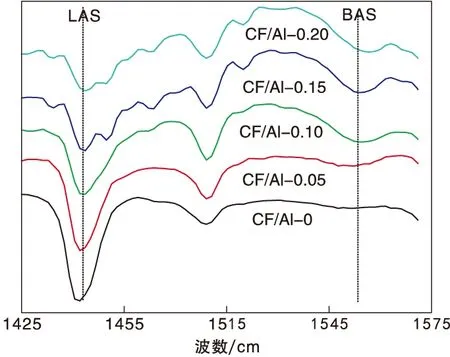
图3 焙烧后氧化铝的吡啶吸附红外光谱图Figure 3 FT-IR spectra of pyridine adsorption on calcined alumina samples

表3 焙烧后改性氧化铝的表面酸性数据
此外,结合表1和表3可以看出,随着掺杂量的增加,B/L值逐渐增加。这可由图4进行解释。研究者在19F MAS NMR光谱中检测到三种类型的氟物质,S1、S2和S3氟物种分别归属于与一个、两个和三个八面体铝原子结合的氟原子。如图4所示,在较低的氟含量下,一个铝羟基被一个氟取代形成S1氟物种。当氟含量过高时,Al-O-Al桥键会断裂,使更多羟基被氟影响[19]。这说明B/L值的突变可归因于S1物种向S2物种转化,使得与氟作用的铝物种成倍增加。虽然掺杂量呈线性增加,但B/L值呈非线性变化,这是L酸随着硼的加入量增加而呈非线性降低引起的[11]。从L酸的变化可知,硼引起的L酸急剧降低也是造成B/L值突变的原因。这说明氟和硼在酸性调变中的协同作用是酸性调变的关键。当F/Al>0.15时,B酸量开始降低。这可能是因为硼不但覆盖L酸位点[20],还会与羟基形成B-O-Al键使原有羟基峰变弱、消失或者转化为新的羟基峰[21],部分作为B酸的羟基也被消耗掉。同时S1物种转化为S2、S3物种,意味着氟含量过高时,大量Al-O-Al键断裂,更多氟进入体相,可极化羟基减少,改性效果也随之降低。
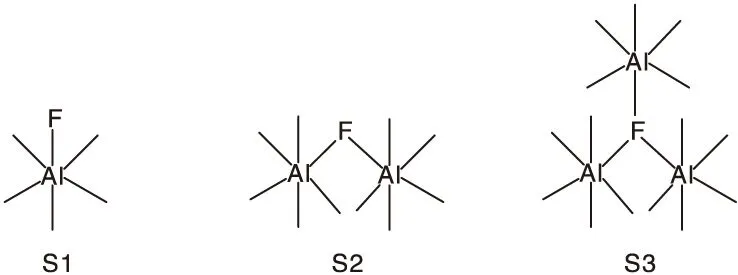
图4 氧化铝表面的不同氟物种Figure 4 Different fluorine species on the surface of alumina
图5为焙烧后改性氧化铝的氮气吸附-脱附等温线及孔径分布图,孔结构及变化率数据见表4。

图5 焙烧后氧化铝的氮气吸附-脱附等温线及孔径分布图Figure 5 Nitrogen adsorption-desorption isotherm and pore size distribution of calcined alumina samples

表4 焙烧后氧化铝的孔结构及变化率数据
从图5可以看出,焙烧后氧化铝的吸附-脱附等温线变化较小,这说明改性对氧化铝孔结构影响较小。由表4可知,改性后样品仍具有较大比表面积,且部分样品孔容和孔径有所增加。在之前的研究中,氟硼酸铵会使改性氧化铝孔结构明显降低[11]。但在本工作中,我们采用浸渍的方法,改性条件更为温和,且拟薄水铝石没有预先胶溶,不会造成氧化铝颗粒的聚集,故孔结构变化较小。
图6为氟掺杂改性氧化铝和以其为载体催化剂的XRD图。

图6 氟掺杂改性氧化铝和以其为载体催化剂的XRD图Figure 6 XRD patterns of F-doped alumina and catalysts supported on them
从图6可以看出,氟掺杂改性氧化铝在2θ=37°,46o,66o附近均出现归属于γ-Al2O3的特征峰,并分别对应于γ-Al2O3的(311),(400)和(440)晶面[22-23]。由改性氧化铝制成的还原态催化剂未出现归属于焦绿石的杂晶峰[11],这主要得益于更低的掺杂量及温和的改性条件。
2.2 富B酸氧化铝的加氢脱硫性能
在常规加氢脱硫反应中,HYD路径与ISO-DDS路径并非完全分开,部分直接脱硫产物可能会进一步加氢或异构化,HYD路径的产物也存在异构化可能性,这会影响路径选择性的判断。因此,在本工作中,使用高压釜作为反应器,通过降低氢气分压抑制产物的深度转化,从而获得转化率低于10%时,初始反应的路径选择性。图7为4,6-DMDBT可能的加氢路径图。一般认为,3,3’-DMBCH、3,3’-DMCHB、4,6-THDMDBT及4,6-HHDMDBT归属于先加氢后脱硫产物;3,3’-DMBP归属于直接脱硫产物;4,4’-DMBP归属于先异构化后直接脱硫产物。如表5所示,因为反应在低氢气分压下进行,所有催化剂均不存在完全加氢产物3,3’-DMBCH。使用商业氧化铝为催化剂,反应后产物以4,6-DMDBT的初步加氢产物4,6-THDMDBT或4,6-HHDMDBT为主,其余产物分别为先加氢后脱硫产物3,3’-DMCHB及直接脱硫产物3,3’-DMBP及异构化脱硫产物4,4’-DMBP,这三种产物均较少且比例相近。这说明对于不含B酸的参比剂而言,低氢气分压使HYD路径被抑制,产物以初步加氢产物为主,脱硫反应无法顺利进行。与文献报道类似,使用含B酸的催化剂时,经DDS或ISO-DDS路径获得的产物比例均有提升。而且通常直接脱硫路径选择性被认为是B酸的作用结果,但当使用分子筛作为B酸来源时,异构化产物中有较多属于裂化产物[24],而使用改性氧化铝作为催化剂时,反应产物中并未检测到裂化产物,这得益于氧化铝具有适宜的酸强度。
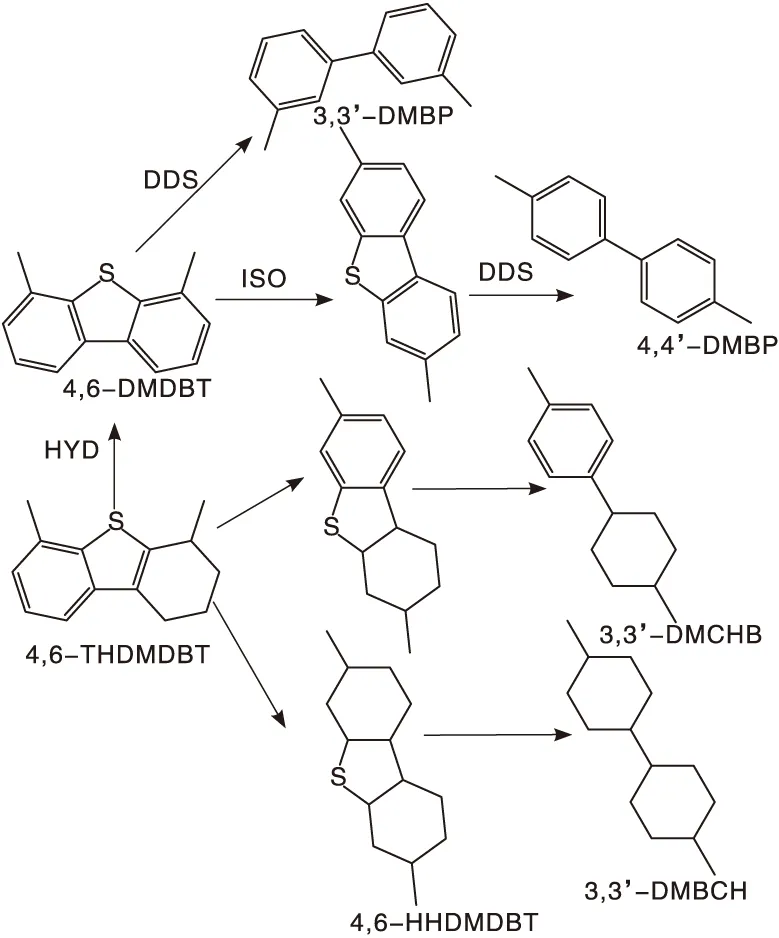
图7 4,6-DMDBT的加氢脱硫路径Figure 7 Hydrodesulfurization path of 4,6-DMDBT
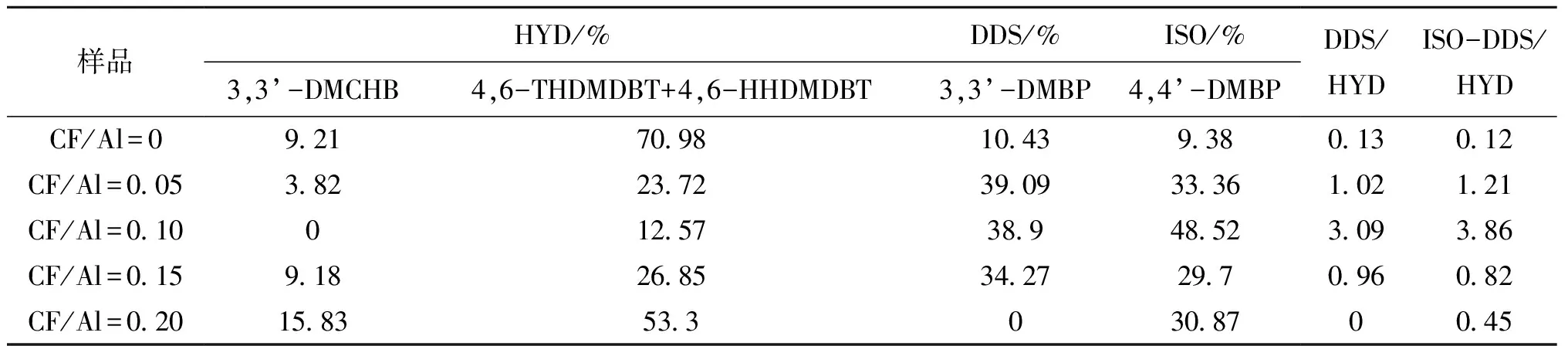
表5 催化剂的加氢路径选择性Table 5 Hydrogenation path selectivity of catalyst
为详细阐述酸性对路径选择性的影响,将B/L值与路径选择性进行关联,结果见图8。由图8可见,随着B/L值增加,DDS/HYD及ISO-DDS/HYD比例均增多,这意味着B酸引入促进了异构化反应的进行,并进一步促进了直接脱硫反应产物的形成,同时也说明B酸使催化剂在相对更低的氢气分压下实现脱硫。但ISO-DDS/HYD比值并未随B酸量线性增加,当B/L值>0.6时,ISO-DDS/HYD比值明显降低。这说明B酸、L酸对脱硫反应具有协同作用,L酸过度降低导致了直接脱硫路径选择性的降低。因为L酸是噻吩类物质吸附的主要活性中心[25],L酸降低不利于噻吩类物质的吸附和活化。

图8 B/L值与DDS/HYD及ISO-DDS/HYD关系图Figure 8 Rrelationship between B/L ratio,DDS/HYD and ISO-DDS/HYD
3 结 论
(1)氟硼酸铵可以对氧化铝的酸性进行较大范围的调变,通过探究掺杂量对氧化铝表面酸性和孔结构的影响,发现未焙烧的改性氧化铝B/L最大可达3.57。虽然焙烧会导致部分氟进入氧化铝体相,在一定程度上减弱改性效果,但改性氧化铝B/L值仍高于1.0。
(2)采用富B酸氧化铝为载体制备的CoMo加氢脱硫催化剂具有更高的4,6-DMDBT脱硫率,且以直接脱硫路径和异构-直接脱硫路径为主。这可归因于富B酸氧化铝具有适宜B/L值,且当B/L值在0.5附近时,DDS/HYD比值及ISO-DDS/HYD比值均达到最大值,分别为3.09和3.86。
